March 13, 2014. Here is a neat trick for combining two separate checkpoint files (.fchk) into a single one. Most useful for combining the forward and reverse separate runs from an IRC calculation.
5 December, 2013. Two new methods for approximately predicting NMR spectrum have been identified. Compared to QM, these are ultra-quick.
2 Dec, 2013. A new option is available for building molecules in 3D (for use in energy minimisation programs such as Avogadro or ChemBio3D). Open ChemDoodle 6 and Structure>Clean>Build 3D Model - This function will generate 3D coordinates, adding hydrogens. Or Structure>Clean>Distance Geometry Embedding - This function is very useful for embedded systems, and will quickly estimate coordinates for structures like adamantane and fullerenes. You can rotate the resulting model in 3D using the Rotate in 3D button (3D cube with red arching arrow, fourth from the left). Export the coordinates in a 3D format (MDL molfile, CML) and import into eg Avogadro for further mechanics minimisation and preparation for Gaussian. ChemDoodle 6 will be deployed on desktop computers in a few days time. See me if you want it installed on laptops. --Rzepa 11:16, 2 December 2013 (UTC)
29 Nov, 2013. A new chemlab1 queue has been created. It uses only 1 processor and runs for a maximum of 1 hour. It should be used for any jobs that can complete within this time, and the purpose is to be able to publish the job to DSpace. Jobs run on a laptop cannot be so published.--Rzepa 14:31, 29 November 2013 (UTC)
25 Nov, 2013. MacBook Air and H: drives. Because the Air can take 2-15 seconds to establish a WiFi connection, it will have completed the login before it does. Under these circumstances, accessing your H: gives an error. To solve this, Finder/Go/Connect to server. Type smb://ic.ac.uk/homes/yourusername and + to remember it in your list of network drives. When you click on it, you should be prompted for your credentials. After this point, H: will become accessible.
15 Nov, 2013. Avogadro2 (for the QTAIM experiment) is now installed on all desktop computers in the two rooms. If you wish it to be installed on your laptop, contact Prof Rzepa. --Rzepa 11:01, 15 November 2013 (UTC)
15 Nov, 2013. Some clarification regarding which alkenes are to be epoxidised. The 1S experiment involves your epoxidising TWO available alkenes from a pool of four (using both catalysts), for which you should compute the NMR and rotation in the 1C experiment.
13 Nov, 2013. Avogadro: New versions have just been received from the developers, which may help with some of the stability issues. Avogadro2 specifically appears much more stable regarding the QTAIM module, which it should be used for in preference to Avogadro1. The latter should be used for other operations. We will try to deploy these programs on a per user basis, so if you want to bring your laptop to me, I will install both programs for you. --Rzepa 07:01, 13 November 2013 (UTC)
21 Oct., 2013. People who obtain their internet from Virgin Media at home may have trouble connecting to the VPN. The fix for this is to log into your router's admin page, and then go to 'Advanced Settings', 'Firewall', tick the box to allow 'PPTP Passthrough', and then click save - you'll be able to log into the VPN after saving that change. Check here if you don't know how to access your router's admin page. --Philip Kent 23:35, 21 October 2013 (BST)
20 Oct.,2013. We have a new Wiki cheatsheet which has been tuned for us! Thanks Tristan!
18 Oct., 2013. All Desktop systems in the downstairs room have now been updated to the latest Java. The desktop computers upstairs will be updated over the coming weekend. The course laptops have also been updated. --Rzepa 12:11, 18 October 2013 (BST)
18 Oct., 2013. URGENT. Following on from the previous message, 8 Desktop computers in the downstairs computer room (along the south edge of the room) are now fully updated and can be used for Wiki editing as needed. More may be added later. --Rzepa 10:35, 18 October 2013 (BST)
18 Oct, 2013. URGENT. Some (all?) of you may have experienced an interesting time getting Java and Jmol to work yesterday. This was caused by the discovery by Oracle of urgent security issues in the version of Java installed on our systems, and hence an urgent need to upgrade. This can only be done by a system administrator, hence the pandemonium. Fortunately, in the last few minutes, a solution has been developed, and we hope to deploy it to as many systems as we can as quickly as we can. Laptops unfortunately can only be updated once they are returned to us. Watch this space for further news.--Rzepa 10:21, 18 October 2013 (BST)
15 Oct, 2013. Computing NMR spectra requires a reference TMS calculated at the same level to be inserted into the Gaussview program. This is done via a file File:Nmr.data which has to reside in the Gaussview folder, sub-directory data. Although all our desktop computers contain the latest version of this file, it is not been updated in the laptop builds. You should download this file and copy it into that folder (you should have admin privs for this operation)
14 Oct., 2013. The HPC job queues contain a mixture of short running jobs (< 30 minutes) along with one potential monster 37 hour effort. This last (the NMR spin-spin couplings for compounds 18 and 17) should be only used if you really feel it will help analyse your NMR; it should not be run just in case. To help avoid building up a large backlog, clogged by a few 37-hour jobs, we have adjusted the maximum run time. Now the Gaussian 4px queue will only run for a maximum of 12 hours (which is more than enough time for all the calculations except the 37-hour monster) and the Gaussian 8px queue will run for 48 hours to allow the monster to complete. So submit all your jobs as 4px, and only run the monster if after careful consideration, you think it will help (for example, if no H-H couplings are reported in the literature, you would have nothing to compare to a calculation). The techniques described here are meant to be generic, and to be useful in other contexts, for example 3rd/4th year research projects. So just because it can be calculated does not mean it should be calculated.
12 Oct., 2013. A reminder that in order to simulate an experimental spectrum, the computed shifts and couplings have to be entered into a simulation program, such as gNMR, available on all computers in the department. Be aware that it is limited in the total number of spins it can handle, probably about 10-12 only.
10 October, 2013. The QTAIM procedure in expt 1C requires Avogadro. Unfortunately, the Windows version of this program has a persistent and currently unresolved crashing bug. The Mac version of this program, as available on the iMac in the level 1 computer room does not (always) crash and hence this unit should be used for this part of the experiment. See also these instructions for how to use Mac Laptops. The Desktop Mac in the computer room is already network-wired and you can ignore those instructions.
To get a screen grab of the QTAIM results, press the cmd, shift (above the ctrl key) and 4 from the numerals at the top. This produces a cross-hair cursor, using which you should drag out the area you wish to capture. When you release this cursor, a screenshot is saved on your desktop, of type .png You can upload this file from the desktop into the Wiki.
The latest version of Avogadro-1.1.1 for Mac can be found here.
A non-crashing Windows version of Avogadro-1.1.1 is not yet available.
6 Oct, 2013. We have made some tweaks during the summer to ensure that the default settings for G09W64 are more appropriate for the resources available on laptops or desktops. In particular, the default memory allocation has been substantially increased, and the number of processors used by default is increased from 1 to 4. This should enable calculations to run (up to) four times as fast than they otherwise would have done. If you observe that what is asserted here is not in fact the case, please contact me. Rzepa 09:02, 7 October 2013 (BST)
Late Breaking news 2012-2013
CRC handbook error. The CRC handbook 93rd edition has an error in it, section 9, page 107, for the stated Thallium - Bromine bond length. The editor-in-chief has been contacted and a correct value of the first digit (before the decimal) was confirmed to be 2, not 1 as stated in the book. --Jrc10 11:14, 18 January 2013 (UTC)
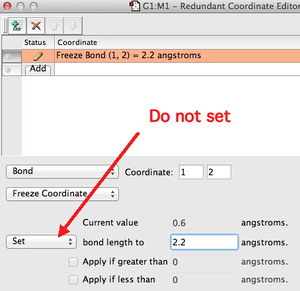
Freezing Coordinates. Finding a transition state can involve specifying a bond as frozen, using the Redundant coordinate editor. Because of a change introduced in Gaussian 09, revision C.01, if you try to set a value for the bond in the editor as shown to the right, when the Gaussian program runs it will ignore this specification completely (resulting in no bond being frozen). So do not use this feature of the redundant coordinate editor.--Rzepa 14:49, 3 December 2012 (UTC)
Referencing a published calculation
You are asked to publish the key calculations you run on the HPC-SCAN system. When you do so successfully, the result will include a field that looks like this:
To quote this result in your wiki, insert this component: {{DOI|10042/21671}} with the result: [1].
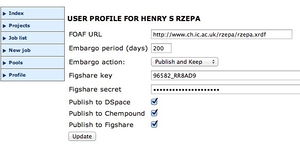
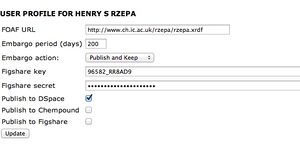
You are using the same system which is normally used to reference scientific publications, but here in the context of a scientific calculation. If you are interested, the full features are shown on the right, and can be set by editing your HPC-SCAN profile. ChemPound is another local repository, and Figshare is an open resource repository which also issues you with a quotable DOI. To publish there however you will need to obtain a Figshare key and secret. The FOAF entry is a so-called RDF declaration, which enables you to identify your credentials for use on the Semantic Web.--Rzepa 08:46, 17 November 2012 (UTC)
The WikED editor. As originally envisaged, the WikEd editor supported Google Chrome, FireFox and Safari. As of 2012 however, the latest Chrome appears to no longer support this editor toolbar. Internet Explorer never has. If you want to take advantage of this editor, please use FireFox only (or Safari on a Mac). There is also another project to improve visual editing, with an expected first release in December 2012.
Using an iPad or iPhone for the course. Much of the course is web browser based. But recently, one can also access the file system to inspect output (.log) files for errors etc. You need to install a (free) app called Mobilecho and configure it as shown here. This allows you to preview files stored in your H: drive (remember to store them there using your laptop or desktop). I have not yet found any IOS app capable of actually viewing the output as a graphically represented molecule, but that may come soon.
Another (this time not free, £2.49) app which might be useful is Wiki-edit, which allows a Wiki to be edited using an iPad.
If anyone discovers functionality for an iPad other than browser and preview based, do let me know. --Rzepa 15:21, 8 November 2012 (UTC)
Avogadro. I an looking for volunteers to try out Avogadro as a replacement for ChemBio3D. The latest version of the program is installed on all laptops and desktops, and its operation is reasonably intuitive. It can serve as a base for performing molecular mechanics pre-minimisation (tidying) of coordinates, and can export these coordinates to other programs such as Gaussian for further analysis.
If you do try to use it, please document your experiences here. --Rzepa 11:20, 7 November 2012 (UTC)
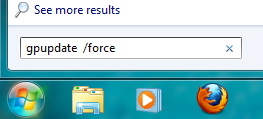
Working with laptops at home. The initial attempt to square the circle by allowing you to open/save files in your H: drive whilst working at home was sub-optimal, with spinning cursor freezes occurring frequently. ICT has taken a close look at the so-called policies configured into the laptop and come up with what is hoped will be a more responsive configuration. To implement this new policy, you have to do the following whilst using the laptop in College (or with the VPN on at home). Type into the search box invoked from the start menu gpupdate /force . This will take around 2-4 minutes. You will need to log out and then in again to complete the process. It is also not unusual to experience spinning cursor delays whilst working in the study area on level 2. This in part is a result of our having to move the location of the laboratory at a late stage into one where the Wireless base station had insufficient capacity. We hope however that a new base station will be fitted very soon which should improve matters in this regard. --Rzepa 12:48, 23 October 2012 (BST)
SCAN job queues. Chemlab 1 job queuesA new job queue has been added. This uses 8-processors in parallel to run a job, whereas the normal queue uses only 4. Please use this new queue for computing larger molecules.
Uploading cub files to create rotatable surfaces on the Wiki. The instructions here describe how you can create a cub file using Gaussview, convert it to a highly compressed format known as .jvxl and upload that to your Wiki. We have established that the cub file must not be in your (network) H: drive if you are using a Windows computer. To be able to upload these files, go to Computer and then drive C:\temp and put all your cub files into the hard drive of the laptop directly. They can then be converted using this link into .jvxl files.
Poor laptop performance at home. There has been a perfect storm of three changes made over the summer: an increase in student H: drive storage quota, procurement of new laptops and a change in appdata policies. It seems that these interact, producing undesirable effects on the laptops when they are taken out of the Campus environment. The new appdata policies in particular seem to be at the centre of things. This means that any application that writes temporary or permanent files now does so to the users H: drive instead of as previously to the local C: drive of the device. This also includes the desktop, in other words any document saved to the desktop is now saved on H: rather than C: (the aim being to ensure that the document is always available to you no matter what machine you sit down in front of). This scheme works well when the laptop is connected to a high bandwidth network, but may not perform so well in a domestic environment. I have asked ICT to look at optimising their policies for laptops so that some of the issues many of you have experienced may be either entirely removed, or at least substantially reduced. These new policies are likely to be tested on Wed 17th October by myself using a standard issue laptop. I will report back here what I discover. So hang on in there! --Rzepa 10:57, 15 October 2012 (BST)
VPN connections and working at home. A number of people have reported that the VNP (virtual private network) connection (a shortcut for which is on the laptop desktop) does not work. I have taken a "vanilla" laptop home and tried to see what happens:
I firstly connected the laptop via an ethernet cable to the Wireless/Broadband router. A network is detected automatically by Windows and configured appropriately.
With a network up, clicking on the VPN desktop icon throws up a prompt to enter the user name, password and domain (the value for the latter should be IC). This worked without problem.
This allows one to e.g. connect to journals and download articles etc (this would not be possible without the VPN working properly)
Note that you do not need the VPN on to log into the Wiki to edit your pages there.
I then pulled the ethernet cable out and used the WiFi connection instead (Control panels/Network and sharing Center), connecting to the SSID of my WiFi router and supplying the security password. ICVPN again connected with no problem, and again I was able to download journal articles, and save into my H: drive. So what might be going wrong that disables the VPN access? Well, one possibility is that the ISP you are signed up with does not support something called VPN Pass-through. VPN is often considered a business feature and many domestic ISP providers have VPN disabled (perhaps as an encouragement for you to sign up with a more expensive business package). Lack of VPN is also often seen with so-called Free WiFi services (often found in hotels and public areas), again possibly to encourage you to sign up for paid-for deals. So the first thing to check would be whether your ISP does actually support VPN. Quite what else might be going wrong would only be speculation on my part. But the bottom line is that the laptops provided for the course ARE capable of VPN connection without any tinkering.--Rzepa 18:01, 11 October 2012 (BST)
Offline H drive. The default setup for undergraduates this year is that the desktop and appdata folders are redirected to the H drive. This can cause mystification for laptop users when the machine is offline. Desktop icons vanish. H:\appdata contains settings for Firefox and other programs and things stop working as expected.
To make things a little easier we have enabled caching for the H drive, so it will be available offline. It will synchronise automatically when you are connected to the college network. Do note that it will also synchronise if you are connected via the VPN and if you are on a capped broadband contract you should bear this in mind. - Nick
Firefox, Java and the Wiki. Activating Java 1Firefox needs Java enabled to run the Jmol applet, which displays molecules, vibrations, MOs etc. The setting which determines this is user specific (it is stored in a file in your H: drive). If you are not getting molecules, but instead a yellow box telling you no Java is enabled, proceed as follows:
In the menus at the top of FireFox, Tools/Add-ons/Plugins.
Scroll down to Java(TM) Platform SE 6 U31 6.0.310.5 and activate it. The security message which indicates that the Java is out of date should be ignored for the time being.
Refresh the Wiki page (or restart the browser). You should get a pop up box which reads The applications digital signature cannot be verified. Run
This indicates that Java is now active again. Run the applet and your molecules should now appear.
Publishing to DSpace. The instructions suggest that you publish select jobs to Dspace. There is current a bug in the profile which prevents this until you have actually activated the publish as shown on the right. Login to the SCAN HPC service and select Profile from the list on the left. Check the button showing Publish to Dspace (it should have been on by default, but in fact its off. Apologies. We hope to fix this shortly).
Also, the publish command needs sometimes to "wake up" the DSpace server. It may not do this the first time you try to publish, but it normally works the second time.
If the job cannot publish, the chances are that there is an error in the job that prevented the writing out of the publication files. You will need to open the job output using Notepad++ (Right-click on the file and select NotePad++ or other text editor such as WordPad) and see what the errors are at the bottom of this file. You may need to discuss these with a demonstrator to determine what went wrong.
Versions of Gaussian. The laptops contain both G03W and G09W 64-bit. Unfortunately, only the first of these is recognised by ChemBio3D V 12. So if you try to run Gaussian from ChemBio3D, it will run G03W. A new release of ChemBio3D V 13 came out over the summer, but try as we might, we cannot get the global licensing codes to reliably work. As an interim measure therefore, we have reverted back to ChemBio3D V12 on your install. However, you can download V13 yourselves for installation on a computer from this site and this will give you a licensing code specific to you which you can use (with only three attempts allowed) to install and license the newer version. G09W contains many updated features, but you would normally be expected to make use of many of them. Perhaps the only significant difference from your point of view is that you can allocate more memory to run Gaussian using the %mem=2GB command. This should make it run (somewhat) faster.--Rzepa 10:56, 5 October 2012 (BST)
Late Breaking news 2011-2012
Incorporating Orbital surfaces into a Wiki
The procedure is as follows
Run a Gaussian calculation on the SCAN
When complete, select Formatted checkpoint file from the output files and download
Double click on the file to load into Gaussview
Edit/MOs and select (= yellow) your required orbitals.
Visualise and Update to generate them
Results/surfaces/contours and from the cubes available list, select one and Cube actions/save cube
Invoke this page and you will be asked to select your cube file,
followed by three file save dialogs, one for the coordinates (.xyz), one for the MO surface (.jvxl) and a package (.jmol).
Insert the following code into your Wiki, replacing the file name with your own choice from the preceding file save dialogs (the string images/4/42/AHB_mo22.jvxl is for illustrative purposes only and must be edited as described below).
Next, upload the the .xyz and .jvxl files into the Wiki (one file at a time, the multiple file uploader does not seem to work for this task)
You will need to find the absolute path for the .jvxl file. Above, this appears as images/4/42/AHB_mo22.jvxl
Just after uploading the .jvxl file, you will see a response as shown on the right.
Right click on the link (blue arrow) and select Copy file location (this can be browser specific)
Paste this string into the above, and edit it down to just images/4/42/AHB_mo22.jvxl (ie trim https://wiki.ch.ic.ac.uk/wiki/ off if that is how it appears)
You should get something akin to:
Orbital
You can superimpose two surfaces. Change the script contents above to
isosurface color orange purple "images/4/42/AHB_mo22.jvxl" translucent;isosurface append color red blue "images/4/42/AHB_mo23.jvxl" translucent;
The four colours used in this line can be changed to whatever you consider appropriate.
An alternative simpler way of loading surfaces
This method avoids the need to specify paths to files as seen above. Instead it uses the .jmol file (as a zip archive) which contains all necessary information and can be invoked by
<jmolFile text="just a link">AHB_mo22.cub.jmol</jmolFile>
which produces just a link. The disadvantage is that it only supports one surface (you cannot superimpose two orbitals). --Rzepa 07:35, 6 March 2012 (UTC)
Report discussion
A reminder that if your report shows a Blue link (rather than red) at the top, you will find some comments on it, which we make during the marking of the report. This is separate feedback to your grade, which is being processed via the Blackboard system, and may appear a day or so after the discussion. So for instant feedback, look at the discussion.--Rzepa 09:51, 1 February 2012 (UTC)
Crystal structure files as initial 3D coordinates for modelling
The start point for obtaining 3D coordinates can be
ChemDraw via its templates feature, which has a wide selection of small molecules, used as an alternative to sketching the molecule from scratch.
The Corina tool, which can be used to create a Molfile of coordinates from a SMILES string generated using ChemDraw.
The Conquest interface to the Cambridge crystal structure database.
Invoke this by typing Conquest into the search box produced by invoking the Windows tool on the bottom left.
When a search is completed, invoke export and Mol2 as the format. Select current entry, one file per entry and save (to desktop).
This file should now be readable using ChemBio3D.--Rzepa 12:45, 20 January 2012 (UTC)
Multiple File uploads
At special request, a multi-file upload facility has been added on an experimental basis. The link appears in the sidebar on the left. Please try out and report any problems --Rzepa 13:48, 27 October 2011 (BST)
DSpace depositions
If you have tried to publish a calculation into Dspace recently, it will have failed. This in part is a knock-on effect from the power outage this last weekend, the full implications of which took several days to sort out. As of this instant, publication is working again. If you have something you tried to publish yesterday (Monday) send the portal Job ID to Mat Harvey (m.j.harvey@imperial.ac.uk) and he will correct the entry.--Rzepa 11:25, 18 October 2011 (BST)
Improvements to the Editing interface
An enhanced editing toolbar called WikEd is now installed for all users and most browsers. You should spend a little while exploring its features.
ChemBio3D version 12
Version 12 (and 11) of this program contain several errors/bugs. The vendors were informed of these more than two years ago. Unfortunately, it seems as if V13 of the program will not be released in time to be used on this course. So you will have to watch out for the bugs.
Late Breaking news 2010-2011
Another way of fixing a broken page
Try this method if the revision history method fails. This is based on trying to back up your project (errors and all) to an editable file,
and then re-editing the file to remove any error that you manange to spot. --Rzepa 21:35, 24 February 2011 (UTC)
Laptop shutdowns at 22.20
Not too many people are actively using their laptop at 22.30, but what has emerged is that routine maintenance procedures are hard-coded (Crontab) into the software build to reboot the system at this time every day. The maintenance involves applying new security patches, new virus definitions, new software releases etc. If your laptop is online just before 22.30, you may notice this activity. Windows 7 then invariably requires the system to be rebooted for the patches to take effect. This will happen irrespective of whether any software patches have been applied. So, at around 22.15 or so, it is essential that you save all work. If you do not, it may be lost when the system reboots. --Rzepa 13:01, 21 February 2011 (UTC)
Mini Project
During the course of grading the project, I discovered some people did have time to do calculations, but the ran out of time for the write up of the project. If you are in such an unfortunate position, do at least put the DOI of the published calculation into your report (this takes just a few seconds). That way, you can at least get some credit for having selected a molecule and calculated it, even if you do not have time to discuss your results on the project page. As with the Wiki report, the DOI entry is also time stamped, so we can easily verify that the calculation was done before the deadline! --Rzepa 09:32, 1 February 2011 (UTC)
Why Wiki
A question occasionally asked is why the course report uses a Wiki instead of the more familiar (to most people) Microsoft word format. Some of the reasons why are summarised here.
Plagiarism
We have implemented strong plagiarism detectors for submitted reports. This would include detecting similarities with previously submitted reports. You should be reminded that plagiarism is taken very seriously indeed, and any detected will have serious consequences for the plagiariser.
Converters for Wiki
It was there, but it got lost (honestly!). See here for some hints on how to convert other formats (Word, HTML) to the Wiki format.
Prettifying Plots produced by Gaussview.
GV (5.09) can visualise spectra (NMR, IT, etc), as well as SCANs and other properties. However, the resulting graphs are not very suitable for inclusion, since the default text sizes are too small, the lines may be very thin and the units for the labels unhelpful. To tweak these spectra, proceed as follows:
Use Gaussview, and select Results/vibrations or NMR/Scan/UV-Vis etc.
Right mouse click and select properties. Here you can select more sensible units, and also the origin.
Right mouse click and select Export. Save the (.svg) file (SVG is to images what HTML is to text).
Using Wordpad (or other text editor), open up the .svg file
Near the top, you will see e.g. the text Scan of Total Energy. Replace by what you want.
You will see font-size:14; Change to something larger.
Further down, you will see font-size:10; Change to something larger.
You will see lots of stroke-dasharray:3; Delete them all (a global replace).
You will see a line starting polyline. It contains stroke-width:0.9; Change to something like stroke-width:2.7;
You will see a line starting rect. It contains rgb(255,255,245); Change this to rgb(255,255,255);
Save the edits, and drop the .svg file onto a FireFox browser Window. The spectrum should appear. If you make further changes, refresh the browser window to see them.
At this point, you probably want to take a screen-grab of the browser window (as a .gif or .jpg file). Upload this to the wiki to incorporate into your report.
Module 2: VPN for home use
Due to the wiki server being hacked it has been brought in behind the college fire-wall. If you want to access it from home (or the scan servers), you must first connect via VPN. If you don't the wiki will not load.
Module 1: Errors in log file
When running SCAN calculations, if no Checkpoint file is produced, or other errors occur, its best to inspect the Gaussian log file using Wordpad. The errors will be seen at the bottom of this file. However, the default font size for Wordpad is 11, which causes the output in the log file to wrap around, causing it to become unreadable. So before you try to inspect this output, set the font size to 9. This produces much more readable outputs.
Module 1: Iodine containing molecules
Although the Mini project is designed to be based on organic molecules, occasionally other elements creep in. One such is iodine, which causes problems because the 6-31G basis set is not available for it. In such circumstances, you can always go to the basis set exchange to get one. A (partial) example of its use is shown below, in which the built in basis is used for C, but the basis set exchange one for I (in fact the 6-311G basis):
# rb3lyp/gen opt
General basis set, including that for iodine
0 1
I -0.58031145 -1.18849341 0.11015250
C 0.73981873 1.76844662 -0.08747886
....
C H etc 0
6-31G(d,p)
****
I 0
S 5 1.00
444750.0000000 0.0008900
66127.0000000 0.0069400
14815.0000000 0.0360900
....
****
Late Breaking news 2009-10
Module 1: Project FAQ
Because the project is defined not in the scripts but by you, it can sometimes seem rather open-ended. So, after an interesting chat with one student, I came up with three possible objectives you could set yourself for the project.
The most ambitious is that you spot a structure in the literature which seems to ring alarm bells. Can it really be that, you ask yourself. Well, if 13C and other spectroscopic data has been reported, you can calculate it and see how well it matches. You might conclude that either its a good match, or not.
A follow up to the first category, is that if the match is poor, you suggest a better one. this is HARD. We do not expect that of you!
More common is that two possible structures have been reported, and you might wish to check that they have been assigned correctly and not transposed.
Another scenario is that the 13C for any given structure is simply reported as a series of values, with no attempt to assign each peak to a specific carbon in the compound. By calculating the spectrum, you can make this assignment.
Any one of the four above is a reasonable objective to set yourself in this project.--Rzepa 15:38, 25 January 2010 (UTC)
Anti-Bredt Natural products?
The blogosphere is buzzing discussing a natural product reported in DOI:10.1016/j.bmcl.2009.10.016 which contains a bridgehead alkene in a small ring. The 13C data is difficult to get from the article, but the structure is an interesting one to analyze using the techniques we show you here. Some of the blogs also are commenting on the early modelling efforts A nice breaking news story that you might wish to look at! (the result of all this attention is a corrigendum, DOI: DOI:10.1016/j.bmcl.2010.04.003 ). And how about this (update: This too has been retracted).
NBO of BH3 has the sAO as the "lone pair"
Something is wrong! The likely problem is that you don't have the ground state lowest energy electronic structure. If you computed the B3LYP/3-21G geometry and then used B3LYP/6-31G to compute the population, the system is not optimised at the B3LYP/6-31G level and your answer will be wrong. Excellent if you noticed this discrepancy. Compute the population analysis and NBO with the 3-21G basis set and you should get a correct analysis. If not, check the energy of your calculation does it give -26.4622632920?
Alternatively if you used the 3-21G basis set ... the new version of G09 prints the NBO analysis slightly differently, the nbo web-page has now been updated to reflect this and explains in detail where to find the correct pz AO.
Could someone perhaps pose the question, to which we see an answer here? --Rzepa 07:32, 20 January 2010 (UTC)
Displaying Vibrations
The write-up section of the course describes how you might animate a vibration using the Wiki. Unfortunately, our upgrade to Gaussian 09 brings with it changes to the manner in which the vibrational information is written out, which Jmol cannot understand. The result is an error message rather than a vibration. I have contact Bob Hanson, editor-in-chief of Jmol and no doubt a fix will be sorted out shortly. --Rzepa 13:09, 16 November 2009 (UTC)
UPDATE: The problem noted above is now fixed. If you take a look at this entry you will see Jmol reading a Gaussian 09 vibration log file and displaying a vibration. This new version of Jmol offers other new features. If you right-mouse-click, a menu appears. The top item of this is the data model. This will contain all the optimisation steps in the log file, together with all the vibrational modes identified. Any one of these can be selected for display.--Rzepa 08:30, 17 November 2009 (UTC)
Backing up your report
Invoke this utility to back your project up. In the box provided, enter e.g. Mod:wzyz1234 being the password for your report. This will generate a page (right) which can be saved using the Firefox File/Save_Page_as menu. Specify Web Page, XML only as the format, and add .xml to the file suffix. You might want to do this eg on a daily basis to secure against corruption. This is in addition to the notes for how to repair broken pages.
We have reports that using the keyword b3lyp/lanl2dz for elements such as Al and Cl produces an error-free log file, but no .fchk file is produced. This is because the conversion program that converts the initially produced .chk file to .fchk (a process needed because the .chk file is specific to the type of computer the calculation was run on, and will not work on Windows computers) is failing to covert the file. We think this is probably an error in the way Gaussian09 produces that .chk file rather than a failure of the converter (the error has been reported to Gaussian for clarification).
One option is to take the optimised geometry and carry out a single point calculation with a full basis set, for example 6-31G(d) which is the same as 6-31G* (just different notation) and then use the checkpoint from this job to visualise the MOs.
Another alternative is to use the following keywords: b3lyp/Gen read=pseudo and then after the blank line that terminates the geometry, to read in the basis set and pseudopotential. These can be obtained from the Basis set exchange. Select all the elements present in your molecule from the Periodic table display, and from the left, select the Orbitals with effective core potential pull down. Then scroll to LANL2DZ ECP, select as the format Gaussian94 and press Get basis set. A Window appears, and you should select all the content in that Window and paste it into your Job input file (using eg Wordpad) after the geometry blank line. One further edit is needed. The section in the basis starting
! Elements ...
is terminated by two blank lines. Delete one of these blank lines (leave the other). The result should look something like this (only Al is shown in part, the other elements are not shown for brevity).
H 3.33099000 -2.03083200 -0.89612200
H 1.96515000 -2.65853800 0.03207800
Al 0
S 2 1.00
0.9615000 -0.5021546
0.1819000 1.2342547
S 1 1.00
0.0657000 1.0000000
P 2 1.00
1.9280000 -0.0712584
0.2013000 1.0162966
P 1 1.00
0.0580000 1.0000000
****
! Elements References
! -------- ----------
! Na - Hg: P. J. Hay and W. R. Wadt, J. Chem. Phys. 82, 270 (1985).
! P. J. Hay and W. R. Wadt, J. Chem. Phys. 82, 284 (1985).
! P. J. Hay and W. R. Wadt, J. Chem. Phys. 82, 299 (1985).
!
AL 0
AL-ECP 2 10
d potential
5
1 304.7291926 -10.0000000
2 61.5299768 -63.8079837
The advantage of the general basis set is that a very much wider selection of basis sets is available (Gaussian only has a sub-set of these built in), including many of the most modern.--Rzepa 09:40, 30 October 2009 (UTC)
Slow Running of laptops
We have had some reports of slow response using Gaussview. This has been identified as having too many programs open simultaneously. This is a generic problem, which we suspect most people experience at some stage in their use of computers. It does not appear to be connected to this lab course in particular. Before you conclude your laptop is malfunctioning, try closing down all the programs except Gaussview and see if it makes a difference.
Associating FireFox with Gaussview 5 not 3
Gausview 5 was received very late by us (late September 2009), and has not been subjected to full testing. We installed it for compatibility with Gaussian 09, and some properties (Vibrations) need GV5 to display properly. Currently, when FireFox is used to inspect Gaussian 09 computed vibrations, it starts up GV3 rather than 5. To reconfigure the browser for GV5, proceed as follows:
In the Tools/Options menu, select Applications
Find the entry for OUT file and Click on the associated action
Set it to always ask
Now a prompt appears on downloading a Logfile, so browse to gauss.exe (in Program files/GV5) and check do this automatically from now on.
This should ensure that GV5 rather than GV3 is invoked automatically.
Queue status
Some people have queried how the queues work. Below is a brief explanation made to those students in an email. It might help you understand what is going on under the hood.
The queues do seem normal in most respects. As of 16.00 there were 16 jobs running in chemlab1 (the max) and 19 pending. By about 20.00 the number of pending jobs were down to 3. This morning at 07.00, only 3 jobs remained in the queues, none pending. This is pretty much what one expects. Chemlab2 on the other hand has all machines still claimed as of this morning. I am checking out if this is simply a full queue, or a hung queue! If the job on this queue does not complete by 7.30, it is stopped (so that the machines can boot back to windows) and an attempt to run it again next night is made. If the job cannot complete in 10 hours (because the molecule is big, ie more than about 20 nonhydrogen atoms), it will only run to completion on weekends, or on chemlab1. This is why the instructions suggest using maxcycles=25 (or less). This limits the job and allows it to complete overnight.
Very much part of this lab is learning to manage your resources. It might be a steep learning curve, but it is also very much what the real research world is about! Fortunately, Moore's law does mean that you get more bang for buck each year. Thus for both vibrations and 13C, Gaussian 09 is about twice as fast as what we had last year! But do remember that the time scales as N**4 (N= number of atoms). So only a few methyl groups extra can double the calculation time!
If individual jobs fail with no output, this probably indicates either a problem with an individual user's account (Matt Harvey will tell us), or that the input is too defective to produce any. In this latter category, do spread the word to come to my office and I will perform a triage on this before escalating to Matt. --Rzepa 07:20, 22 October 2009 (BST)
Remote connections and use of Gaussview on non College Computers
RDC connection
With laptop hand-back not fully overlapping with write-up deadlines, a number of people have asked how they might be able to access the course programs without the need to physically be present in the two computer rooms. The Gaussview license in particular does not allow installation on non-Imperial computers. To overcome this restriction, you can do the following:
Use Remote Desktop Connection, which is installed on most Windows computers.
Establish a VPN connection if you are off-campus
Connect to chas.ch.ic.ac.uk (the chemistry application server). You will be presented with the same screen as you would if you were physically at the computer. This includes all the software etc.
You will find that some operations are more stodgy than others (surface rendering for example) but if you have a reasonably fast (i.e. broadband) connection, the experience is not too bad.##You can test the above (but without the need to set up the VPN) from one of the static computers in the computer rooms.--Rzepa 12:55, 18 December 2008 (UTC)
Conversion of Word to Wiki format
If you prefer to author your report using Word, and then at the final stage convert it to the Wiki, you can do so using OpenOffice 3.0. This has a Word to MediaWiki converter which allows you to save the file in Wiki text. This can then simply be pasted into your Wiki page. It might need some tidying up (in particular, Jmol molecules can only be added at the wiki stage, and not so in Word itself), and you will have to still upload the graphics in the Wiki. Since Januar 2009, V 3.0 of openoffice has been installed which supports this feature.--Rzepa 17:05, 23 January 2009 (UTC)
Several people have reported that Word is crashing during use. We think it might be because you have used it to open a document located on the network drive H:. If your network goes down (see above) whilst you are editing the document, Word may well panic and hence crash! To avoid this, and whilst you still have a network, make a copy of any Word document from drive H: to the hard drive E:. Then, only ever edit that local copy. Finally, when you are finished, copy the document back from E: to H:. Hopefully, this will avoid any Word crashes.--Rzepa 10:37, 17 October 2008 (BST)
Email Alerts
You can receive email alerts if any page on the wiki is changed (ie when additions to this page are made). Click on the watch link at the top of any page and then on the my preferences link (you have to be logged in), tick the E-mail me when a page I'm watching is changed box.