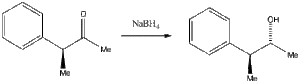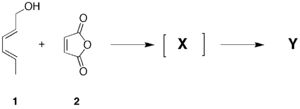Please feel free to try these problems in your own time, and to discuss these with your organic tutors and lecturers. Note also that the relevant lectures may occur in the spring as well as autumn terms.
Axial/Equatorial preferences in cyclohexane and cyclohexanone and Hydrogen Bonding
Cyclohexanone
Construct a chair cyclohexane and replace firstly one of the axial hydrogens with the following groups: methyl, t-butyl, OH. Calculate the energy of the axial isomer.
Then repeat (either by deleting/redrawing or by moving) for the equatorial forms. Compare the energies of the two isomers. Does any energy difference increase with the size of the group? Does OH fit into this in terms of size?
thiomethyl cyclohexanoneThe dissolving metal reduction of cyclohexanones in a protic solvent (i.e. one capable of hydrogen bonding) is thermodynamically controlled and gives the more stable, equatorial alcohol. In fact, its probably the alkoxide that is the product, not the free alcohol. It is thought the alkoxide is actually a lot larger than the alcohol, accounting for the substantial equatorial preference. Can you think why its larger? [Ghemical cannot in fact model this, since the force field does not include parameters for the alkoxide anion].
Determine the axial/equatorial preference of 2-methylthio-cyclohexanone (Hint: there are many conformations possible, and you should try a few to see if you can get the lowest).
References and Footnotes
A. H. Lewin and S. Winstein, NMR. Spectra and Conformational Analysis of 4-AlkylcyclohexanolsJ. Am. Chem. Soc.; 1962, 84, 2464 - 2465; DOI:10.1021/ja00871a049
F. R. Jensen and L. H. Gale, The Conformational Preference of the Bromo and Methyl Groups in Cyclohexane by IR Spectral Analysis, J. Org. Chem., 1960, 25, 2075 - 2078. DOI:10.1021/jo01082a001
K. B. Wiberg, J. D. Hammer, H. Castejon, W. F. Bailey, E. L. DeLeon, and R. M. Jarret, Conformational Studies in the Cyclohexane Series. 1. Experimental and Computational Investigation of Methyl, Ethyl, Isopropyl, and tert-Butylcyclohexanes, J. Org. Chem., 1999, 64, 2085 - 2095; DOI:10.1021/jo990056f . The salient point here is that the enthalpy andentropy of this series differ in their trends.
Just when you are starting to think that things are quite simple, along comes the observation: S. E. Biali, Axial monoalkyl cyclohexanes, J. Org. Chem., 1992, 57, 2979 -2980; DOI:10.1021/jo00037a001
And this one with knobs on: In all-trans-1,2,3,4,5,6-hexaisopropylcyclohexane, all the alkyl groups are located at axial rather than equatorial positions: O. Golan, Z. Goren, and S. E. Biali, Axial-equatorial stability reversal in all-trans-polyalkylcyclohexanes, J. Am. Chem. Soc., 1990, 112, 9300 - 9307. DOI:10.1021/ja00181a036 .
J. A. Anderson, K. Crager, Kelly, L.Fedoroff, G. S. Tschumper, Gregory S. Anchoring the potential energy surface of the cyclic water trimer.J. Chem. Physics, 2004, 121, 11023-11029. DOI:10.1063/1.1799931 .
R. R. Fraser, N. C. Faibish, On the purported axial preference in 2-methylthio- and 2-methoxycyclohexanones: steric effects versus orbital interactions, Can. J. Chem., 1995, 73, 88-94.
Conformational analysis II: cis and trans-decalins
Elimination
Woodward
A key step in Woodward's famous synthesis of [1] is a quinone+butadiene Diels-Alder reaction to give a cis-decalin (left), with an assumption that epimerisation to a trans-decalin is thermodynamically favourable.cis CortisoneCan you verify whether the trans-isomer is indeed more stable? Its not so obvious, since this compound has two extra double bonds in the rings and six sp2 centres which might perturb things.
trans Decalin The two diastereomeric trans-decalin tosylates react quite differently with NaBH4. Construct models for both isomers (use methoxy as a model for the Tosyl group) and from the antiperiplanar alignments of bonds that you can find in each isomer, can you make a connection to the reactivity of each form? Consider very carefully where you would put a lone pair located on the nitrogen (i.e. include the N-Lp "bond" in your antiperiplanar alignments) asuming the this atom is tetrahedral rather than planar. Does this lone pair play any part in either reaction in this position?. Note that the relative energy of the axial/equatorial N-Methyl group will not be an accurate reflection of any antiperiplanaralignments, since these are predominantly electronic in origin, and this mechanics method does not take these into account.
The second (elimination) reaction is very slow compared to the first. Discuss with tutors why this might be so (for Hints, see here or here).
These reactions do not appear to occur for the corresponding cis-decalins6. Why not?
For a modern application of mechanics to this molecule, see J. M. A. Baas, B. Van de Graaf, D. Tavernier, and P. Vanhee, Empirical force field calculations. 10. Conformational analysis of cis-decalin, J. Am. Chem. Soc., 1981, 103, 5014 - 5021;DOI:10.1021/ja00407a007 .
For a video-Podcast of Barton and Woodward (and other Nobel prize winners), subscribehere
R. B. Woodward, F. Sondheimer, and D. Taub, The total Synthesis of Cortisone, J. Am. Chem. Soc., 1951, 73, 4057 - 4057. DOI:10.1021/ja01152a551 .
P.-W. Phuan and M. C. Kozlowski, Control of the Conformational Equilibria in Aza-cis-Decalins: Structural Modification, Solvation, and Metal Chelation, J. Org. Chem., 2002, 67, 6339 - 6346; DOI:10.1021/jo025544t
Thermodynamic vs Kinetic Control Part 2.
Menthone
Enols The molecule on the right has four different hydrogens that could potentially be removed by treating the compound with base. Construct the model, and from inspection, can you decide which of the four possible enols might form? Calculate the energies of all four. Which is lowest? (Hint: the carbanion orbital resulting from proton removal has to be able to conjugate with the π-system of the carbonyl group).
How to induce room temperature hydrolysis of a peptide
Pentahelicene
Peptide hydrolysis This introduces a further example of how simple conformational analysis can quickly rationalize kinetic behaviour. At neutral pH and 25° the half life for hydrolysis of a peptide bond is around 500 years (and thank goodness, or we would ourselves all rapidly hydrolise to a mush!). Some enzymes however can achieve this in less than 1 second, an acceleration of 1013! Organic chemists are not quite so clever, but they can achieve room temperature hydrolysis of a peptide in 21 minutes by careful conformational design. The two isomers shown on the right differ only in their stereochemistry, one hydrolysing quickly, the other slowly. Build a model of each compound, and calculate two isomers for each, varying in whether the ring N-substituent is oriented axial or equatorial with respect to the decalin ring. On the basis of your two pairs of energies, can you rationalise the observed kinetic behaviour? Do you know why both of these compounds take very much less than 500 years to hydrolise the peptide bond?
Hint1: Use the chair-chair conformation for cis-decalin as your template for constructing this system.
Hint2: When constructing your models, think if there are any hydrogen bonds that might stabilize the structure!
Hint3: Hydrolysis can only occur when the OH group can approach the carbonyl of the peptide bond close enough to react, and at the right angle of approach.
Reference
M. Fernandes, F. Fache, M. Rosen, P.-L. Nguyen, and D. E. Hansen, 'Rapid Cleavage of Unactivated, Unstrained Amide Bonds at Neutral pH', J. Org. Chem.,2008, ASAP:DOI:10.1021/jo800706y
Caryophyllene: The phenomenon of Atropisomerism
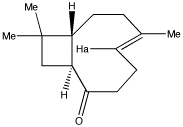
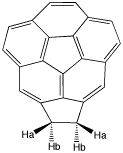
Caryophyllene ketoneCaryophyllene, a constituent of many essential oils, include clove oil, has atrans alkene contained in a 9-membered ring. One interesting property is that it has 4 diastereoisomers possible, originating from a total of three asymmetric centres present in the molecule. Two of these are conventional chiral centres, one is present in the form of a disymmetric trans double bond. To understand why such a bond can result in two configurations, one must appreciate that (concurrent) rotation about the two C-C single bonds adjacent to the alkene is in fact restricted, because to the hydrogen labelled Ha cannot easily pass by the edge of the 4-membered ring. Construct this molecule (in fact the ketone rather than the alkene) and optimize its geometry. Note in particular that the ring junction is trans and not cis.
You will find you may well have obtained one of two forms. In the first, the Ha hydrogen will be opposite the C=O group, in the other it will be adjacent to it. Record the energy of whatever form you got. At the end of the course, we will try to find the winner with the lowest energy (this is not as trivial as it sounds!).
Next, take your structure, and try to flip the trans alkene bond around so that eg if the methyl were previously pointing up, now it will point down. You may find a combination of erasing/redrawing or of moving, will accomplish this. You may also find another trick useful, of deleting all hydrogens, and then re-sprouting them back on again. Re-optimise your structure and compare the energy with your first isomer.
Another feature of this model is that you can judge which group is in the so-called shielded region of the carbonyl group magnetic anisotropy. Using this information, you can see if there are any anomalous 1H chemical shifts that might need explaining!
References
M. Clericuzio, G. Alagona, C. Ghio, and L. Toma, Ab Initio and Density Functional Evaluations of the Molecular Conformations of -Caryophyllene and 6-Hydroxycaryophyllene, J. Org. Chem.2000, 65, 6910 - 6916. DOI:10.1021/jo000404+ .
For a recent application of this phenomenon, see P. C. Bulman Page, B. R. Buckley, S. D.R. Christie, M. Edgar, A. M. Poulton, M. R.J. Elsegood and V. McKee, A new paradigm in N-heterocyclic carbenoid ligands, J. Organometallic Chem., 2005, 690, 6210-6216. D DOI:10.1016/j.jorganchem.2005.09.015 .
Germacrene: Conformational analysis of medium sized rings
Germacrene
Germacrene and the thermal reaction productGermacrene is a natural product with a ten-membered ring; it has the triene structure shown. Assuming that it adopts a crown conformation, build a three-dimensional model.
On heating, germacrene is converted into one of the stereoisomers of the divinylcyclohexane, via a [3,3] sigmatropic pericyclic reaction. Predict from your model for Germacrene whether the product will have the two vinyl groups cis or trans to one another.
References
K. Shimazaki, M. Mori, K. Okada, T. Chuman, H. Goto, K. Sakakibara and M. Hirota, Conformational analyses of periplanone analogs by molecular mechanics calculations, J. Chem. Ecology, 1991, 17, 779-88. DOI:10.1007/BF00994200 .
H. Shirahama, E. Sawa and T. Matsumoto, Conformational aspects of germacrene B. Are the germacrenes resolvable ?, Tetrahedron Lett., 1979, 20, 2245-2246. DOI:10.1016/S0040-4039(01)93687-1 . See also DOI:10.1039/P19750002332 for an explanation of the selective epoxidation of germacrene.
Xestoquinone: Regio and Stereoselectivity in the Diels Alder reaction
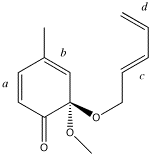
Xestoquinone precursor This compound is a precursor to a natural product called Xestoquinone. It has four alkene groups, which can individually be considered as the alkene component in a π2s + π4sDiels Aldercycloaddition. The pair of alkenes a+b or c+d can also act as the diene component in the π2s + π4sDiels Aldercycloaddition. Construct a model of the product of e.g. forming a bond between alkenea or alkene b and diene c+d, and then reverse the addition by using either c or d adding to the diene a+b. The stereochemistry of addition should always be suprafacial, i.e. preserving the stereochemical relationships of the alkenes. You should very carefully check that this is so in your final model.
Whilst you should stop at two models, it is possible to construct many more. For example, one might be able to add to either the top face of alkene b or to its bottomface. Identify the model with the lower energy, and save it for the end of the workshop. We will identify the isomer of lowest energy from everyone's results, this being a communal Monte Carlo experiment to find the global minimum.
For the original literature on this synthesis, see R. Carlini, K. Higgs, C. Older, S. Randhawa, and R. Rodrigo, Intramolecular Diels-Alder and Cope Reactions of o-Quinonoid Monoketals and Their Adducts: Efficient Syntheses of (±)-Xestoquinone and Heterocycles Related to Viridin, J. Org. Chem., 1997, 62, 2330 - 2331. DOI:10.1021/jo970394l where you can check to see which isomers actually do form!
Aldol Reaction and anti-Bredt Rings
Aldol
Aldol ReactionWhen the diketone shown is treated with base, it undergoes an aldol condensation. Two obvious possibililties are elimination of the combination Ha and Oa, or of the alternative combination Hb and Ob. In fact, only a single product is formed. On the basis of energies for both products, can you predict which one is actually formed?
Measure a few dihedral angles, ie to find out how planar the alkene present is. Does this suggest a reason why one isomer is less stable than the other?
There is a third very remote structural possibility. If you have time, verify that this third product truly is unlikely.
I. Novak, Molecular Modeling of Anti-Bredt Compounds, J. Chem. Inf. Model., 2005, 45, 334 - 338. DOI:10.1021/ci0497354
See also this article A. Nickon, D. F. Covey, F.-C. Huang, and Y.-N. Kuo, Unusually facile bridgehead enolization. Locked boat forms in anti-Bredt olefins, J. Am. Chem. Soc., 1975, 97, 904 - 905; DOI:10.1021/ja00837a043 in conjunction with Project 9.
Conformational Preference for asymmetric hydride reduction of a ketone
Asymmetric hydride reductionThe hydride (BH4, AlH4, etc) reduction of the ketone shown here is stereospecific, resulting in an alcohol with the stereochemistry shown (known as the Cram or the Felkin-Anh rule). Construct a model of the ketone and establish which of at least two conformations is the lowest in energy.
If the hydride anion is delivered from the least hindered position, is the conformation you have consistent with the stereochemistry shown for the product?
You can see from Ref 4 that the situation can be far more complex, depending on many other factors.
D. J. Cram and D. R. Wilson, Studies in Stereochemistry. XXXII. Models for 1,2-Asymmetric Induction, J. Am. Chem. Soc., 1963, 85, 1245 - 1249. DOI:10.1021/ja00892a008 .
Y. Yamamoto, K. Matsuoka, and H. Nemoto, Anti-Cram selective reduction of acyclic ketones via electron transfer initiated processes, J. Am. Chem. Soc., 1988, 110, 4475 -4476; DOI:10.1021/ja00221a093 .
A. Mengel and O. Reiser, Around and beyond Cram's Rule, Chem. Rev., 1999, 99, 1191 - 1224. DOI:10.1021/cr980379w .
Enantiomers vs Diastereomers Part 4: NMR Coupling constants
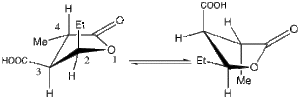
Axial-equatorial interconversionIn Project 2.2 above, we saw how the energies of diastereomeric compounds could be compared with the corresponding enantiomers. In this extension, we show how molecular modelling can cast light on the conformation adopted by 2-ethyl-4-methyl-1-oxa-cyclopentane-3-carboxylic acid estimated using measured1H NMR coupling constants. The (2S,3S,4S) diastereomer has couplings of 3JH2,H3 8.3 Hz and 3JH3,H4 9.8 Hz. Two possible conformations of this diastereomer are shown on the right. They differ in that one has Et axial, and Me/COOH equatorial, and the other Et equatorial and Me/COOH axial.
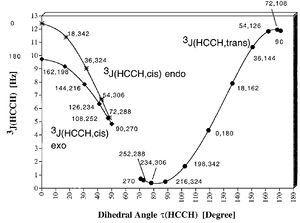
Karplus plot. Click to expandBy calculating the geometries of both conformations, and measuring the dihedral angle H2-C-C-H3 and H3-C-C-H4, one can assess by using the Karplus equation (left, taken from Ref 2 and relevant for a cyclopentane, but the values for which might be modified by the presence of electronegative substituents), which conformation leads to the best agreement between the calculated angle and the measured coupling constants (Hint: on the basis of the predicted couplings, you should be able to eliminate one of the two conformations shown for this molecule).
5-circuleneIn Project 2.2 we also introduced molecules such as helicenes and circulenes. The 1H NMR of the [5]-circulene shown to the right revealed a complex spectrum at δ 2.98 ppm and again at 3.75 ppm. On the face of it, the four protons labeled Ha and Hb should all be equivalent, and the spectrum should be a single peak, not two complex multiplets. Indeed, if the NMR is recorded at high temperatures, this is exactly what is observed. By constructing a model of the [5]-circulene shown, can you explain why at normal temperatures, the NMR spectrum is so complex?
Synthesis lab experimentA practical application of this technique is to determine the stereochemistry of the product of the reaction between E,E-2,4-hexadien-1-ol and maleic anhydride. You will have the 1H NMR spectrum of your sample recorded, and evident from that will be peak multiplicities of the various proton resonances. You should endeavour from your analysis to come up with a suggestion for the structure of compound Y, and from this, estimates of the numerical values (but not the signs) of the 2J and 3J couplings visible. Now using the techniques described above, construct a model of your proposed structure for Y. Measure the dihedral angles for all the 3J couplings, and very approximately estimate what the corresponding 3J might be from the diagram above. Does this help you assign the stereochemistry of the product?
Advanced topic: Part of the spectroscopic analysis of the compound Y involves interpreting the IR spectrum. Theory can be used in fact to simulate the full IR spectrum. In section 5.3 below, you will find instructions on how to use the model you have calculated here to initiate a so called density functional calculation. This will provide you with the required IR simulation. Follow these instructions, and open the resulting .log file in Gaussview. Go to the Results menu and select vibrations. The IR spectrum will be displayed. Does it match the one you have recorded for yourself?
References
M. Karplus, Vicinal Proton Coupling in Nuclear Magnetic Resonance, J. Am. Chem. Soc., 1963, 85, 2870 - 2871; DOI:10.1021/ja00901a059
A. Wu, D. Cremer, A. A. Auer, and J. Gauss, Extension of the Karplus Relationship for NMR Spin-Spin Coupling Constants to Nonplanar Ring Systems: Pseudorotation of Cyclopentane, J. Phys. Chem. A,, 2002, 106, 657 -667; DOI:10.1021/jp013160l
C. A. Stortz and M. S. Maier, Configurational assignments of diastereomeric γ-lactones using vicinal H–H NMR coupling constants and molecular modelling, J. Chem. Soc., Perkin Trans. 2, 2000, 1832 - 1836. DOI:10.1039/b003862h
A. H. Abdourazak, A. Sygula, and P. W. Rabideau Locking the bowl-shaped geometry of corannulene: cyclopentacorannulene, J. Am. Chem. Soc., 1993, 115, 3010 - 3011.DOI:10.1021/ja00060a073
Bridgehead enols: Thermodynamic vs Kinetic Control Part 2.
Bridgehead
Brendanone The ketone Brendan-2-one shown right exhibits unusual behaviour.6 When treated with NaOD/MeOD, deuterium substitution occurs easily and rapidly only in position Hb. Enolisation must of necessity form a bridgehead double bond (anti-Bredt), but clearly one isomer is more stable than the other possible form. Does molecular modelling predict this correctly?
The unusually facile enolisation of this ketone (given that it forms an anti-Bredt enol) can also be investigated by molecular modelling. Measure the dihedral angle between the C-Ha or C-Hb vector and the carbonyl group. Assuming that the ideal angle for proton removal is around 90°, which proton is better set up for abstraction? Might this be kinetic rather than thermodynamic control?
CortisoneOne could also revisit Problem 2.3.3 above. Here, proton abstraction forms an enol which eventually epimerises the bridgehead position to form atrans ring junction. Why should this proton be particularly easy to remove? From what you have learnt above, would this be for kinetic or for thermodynamic reasons (or both?). Are all the relevant effects modelled using the mechanics approach or is consideration of the electrons also necessary?
Sulfonylation of Naphthalene: Thermodynamic vs Kinetic Control Part 3.
Sulfonylation of naphthalene
The sulfonylation of naphthalene using sulfuric acid is a good example of a mechanism combining both steric and electronic influences. The Molecular mechanics method intrinsic to the Ghemical program can only model the former, and not the latter. It is a worthwhile exercise to establish whether this anticipated deficiency does indeed lead to a model which only partially explains experiment.
It has been known for some time that treating naphthalene with sulfuric acids at low temperatures produces mostly substitution at the 1-position of the naphthalene. Heating the reaction mixture, or conducting the reaction at elevated temperatures produces mostly the 2-isomer. This is indeed a classic example of kinetic vs thermodynamic control, the 1-isomer being the kinetic one and the 2-isomer the thermodynamic one. To model the kinetic reaction, we have to inspect the transition state for the reaction, and here we can approximate this by the Wheland Intermediate. To model the thermodynamic reaction, we have to inspect the product (rather than the transition state) for the reaction.
Build models for all four species shown in the diagram on the right. For the two products, define conjugated bond types for all the ring bonds, and define the sulfonyl group with two S=O double bonds and one S-O single bond. Take care to optimise the conformation of the sulfonyl group with respect to the aromatic ring. For the two Wheland intermediates, the limitations of Ghemical will force us to cheat. Ghemical does not have parameters for a carbocation. So define the C2-C3 bond as conjugated (for the 1-Wheland intermediate). When you add hydrogens it will in fact add a second hydrogen to C2. Delete this one hydrogen. Ghemical will calculated the energy regardless of not knowing C2 is actually a carbonium ion! For the 2-Wheland intermediate, ensure that you use exactly the same number of conjugated bond types as you did for the 1-isomer (the two models in a mechanics sense are only comparable if you have the same total number of bond types in each model). You will have to decide whether these (undoubted) approximations have produced reasonable models or not (is the naphthalene framework planar for example, as it should be?).
Record the pairs of energies (two for the 1- and 2-products, and two for each preceeding transition (Wheland) state.
By turning the spacefilling representation on, which of the two products has the least unfavourable steric interactions between the sulfonic acid group and any adjacent hydrogens? Does this match with their relative energies?
Do any unfavourable steric interactions observed in the product(s) also exist in the Wheland intermediates (as models for the transition states)?
The relative stability of the Wheland intermediates is always assumed to be an electronic phenomenon. The conventional explanation is that the 1-Wheland isomer is stablized by both one aromatic ring and an allyl cation conjugated to it. The 2-Wheland isomer is stabilised by one aromatic ring conjugated to a secondary carbocation and an alkene. This type of cross conjugation is conventionally assumed to be less favourable. Does a purely mechanical approach to this problem reproduce this expectation? Or is this mechanical approximation to anelectronic model too severe? It seems a good point to stop this course, since the next time you will build models, it will indeed be using methods which properly approximate the electronic components.
References
R. Lantz, Mechanism of the monosulfonation of naphthalene, Compt. Rend. 1935, 201, 149-52.
G. W. Wheland, A Quantum Mechanical Investigation of the Orientation of Substituents in Aromatic Molecules, J. Am. Chem. Soc.1942, 64, 900 - 908; DOI:10.1021/ja01256a047
C. A. Reed, N. L. P. Fackler, K-C. Kim, D. Stasko, D. R. Evans, P. D. W. Boyd, and C. E. F. Rickard, Isolation of Protonated Arenes (Wheland Intermediates) with BArF and Carborane Anions. A Novel Crystalline Superacid, J. Am. Chem. Soc.1999, 121, 6314 - 6315 DOI:10.1021/ja981861z