
Rep:Mod:fred234
Physical Computational Chemistry
Introduction
This computational experiment is about characterising the transition structures for the Cope rearrangement and Diels Alder cycloaddition reactions. In this experiment, the transition structures in larger molecules (such as the transition structure of orto-thalic acid anhydride) were shown and studied. The Schrodinger equation was solved numerically, using the molecular orbital-based method, by locating the transition state structures (based on the local shape of the potential energy surface). Reaction paths and activation energies were also be calculated in this experiment.
Hart-tree optimisation
1,5-hexadiene (anti)
The total energy for the "anti" linkage of 1,5-hexadiene is -231.69253 a.u. and the point group for the structure is Ci.

This is the Hart-Tree optimised 1,5-hexadiene (anti) file: opt_anti.
Summary table
| Summary | Results |
|---|---|
| File name | 1,5-hexadiene_optimisation_anti2 |
| File type | .log |
| Calculation Type | FOPT |
| Calculation Method | RHF |
| Basis set | 3-21G |
| Charge | 0 |
| Spin | Singlet |
| E(RHF) | -231.69254 a.u. |
| RMS Gradient Norm | 0.00003 a.u. |
| Imaginary Freq | |
| Dipole moment | 0.00 Debye |
| Point Group | Ci |
1,5-hexadiene (gauche)
The gauche conformation should be higher in energy because the hydrogens are nearer to each other in the gauche confromation - this means that the hydrogens are in a sterically less favourable position in this conformation. This is indeed the case; the total energy for the gauche conformation is -231.68916 a.u. (in comparison to -231.69254 for the - previous - anti-conformer). The point group for the gauche conformer is C1.

This is the Hart-Tree optimised 1,5-hexadiene (gauche) file: opt_gauche.
Summary table
| Summary | Results |
|---|---|
| File name | 1,5-hexadiene_optimisation_gauche |
| File type | .log |
| Calculation Type | FOPT |
| Calculation Method | RHF |
| Basis set | 3-21G |
| Charge | 0 |
| Spin | Singlet |
| E(RHF) | -231.68916 a.u. |
| RMS Gradient Norm | 0.00000 a.u. |
| Imaginary Freq | |
| Dipole moment | 0.54 Debye |
| Point Group | C1 |
1,5-hexadiene (anti)

This is the Hart-Tree optimised 1,5-hexadiene (anti) file: opt_anti_new.
This anti conformer has a total energy of -231.69097 a.u. and a point group of C1.
Summary table
| Summary | Results |
|---|---|
| File name | 1,5-hexadiene_optimisation_anti |
| File type | .log |
| Calculation Type | FOPT |
| Calculation Method | RHF |
| Basis set | 3-21G |
| Charge | 0 |
| Spin | Singlet |
| E(RHF) | -231.69097 a.u. |
| RMS Gradient Norm | 0.00001 a.u. |
| Imaginary Freq | |
| Dipole moment | 0.30 Debye |
| Point Group | C1 |
Lowest energy conformer of 1,5-hexadiene
The lowest energy conformer will have the two ethene groups anti-periplanar with respect to each other. The total energy of the lowest energy conformer is -231.6926 a.u. and the point group of the structure is C2.
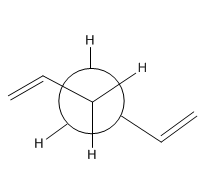

This is the Hart-Tree optimised lowest energy conformer of 1,5-hexadiene file: opt_lec.
Summary table
| Summary | Results |
|---|---|
| File name | 1,5-hexadiene_optimisation_anti1 |
| File type | .log |
| Calculation Type | FOPT |
| Calculation Method | RHF |
| Basis set | 3-21G |
| Charge | 0 |
| Spin | Singlet |
| E(RHF) | -231.69260 a.u. |
| RMS Gradient Norm | 0.00002 a.u. |
| Imaginary Freq | |
| Dipole moment | 0.20 Debye |
| Point Group | C2 |
DFT optimisation of anti2

The basis set for the (DFT) optimisation was changed from 3-21G to B3LYP/6-31G(d). The total energy changed from -231.69254 a.u. to -234.61171 a.u.. The point group for the two anti2 structures is still Ci, but the structure that was derived with higher level of theory is lower. The (tetrahedral) angle is 109.40 degrees, for the Hart-tree optimised anti2 conformer, and 109.61 degrees, for the DFT optimised conformer; the larger tetrahedral angle for the DFT optimised anti2 conformer is the reason for why it calculates a lower energy (there's less steric repulsion).
This is the DFT optimised anti2 conformer of 1,5-hexadiene file: opt_anti2.
This is the frequency file: freq_anti2.
Summary table
| Summary | Results |
|---|---|
| File name | 1,5-hexadiene_frequency_anti2_B3LYP6-31Gd |
| File type | .log |
| Calculation Type | FREQ |
| Calculation Method | RB3LYP |
| Basis set | 6-31G(d) |
| Charge | 0 |
| Spin | Singlet |
| E(RB3LYP) | -234.61171 a.u. |
| RMS Gradient Norm | 0.00000 a.u. |
| Imaginary Freq | |
| Dipole moment | 0.00 Debye |
| Point Group | Ci |
Comparison with appendix
The anti-periplanar conformer with a total energy of -231.69254 a.u. is the anti2 conformer; the anti-periplanar conformer with a total energy of 231.69260(236) a.u. is the anti1 conformer; the gauche conformer shown is the gauche6 conformer; and the anti-periplanar conformer with a total energy of -231.69097 a.u. is the anti4 conformer.
Low frequencies
Low frequencies --- -9.2018 -0.0006 -0.0005 -0.0005 3.4135 13.2274 Low frequencies --- 74.4382 81.0434 121.4893
Item table
Item Value Threshold Converged?
Maximum Force 0.000019 0.000450 YES
RMS Force 0.000006 0.000300 YES
Maximum Displacement 0.000239 0.001800 YES
RMS Displacement 0.000076 0.001200 YES
Predicted change in Energy=-3.992139D-09
Optimization completed.
-- Stationary point found.
IR spectrum

Thermochemistry at 298.15 K
Sum of electronic and zero-point Energies= -234.469203 (Hartree/Particle) Sum of electronic and thermal Energies= -234.461858(Hartree/Particle) Sum of electronic and thermal Enthalpies= -234.460913(Hartree/Particle) Sum of electronic and thermal Free Energies= -234.500775(Hartree/Particle)
Thermochemistry at 0 K
Sum of electronic and zero-point Energies= -234.469203 (Hartree/Particle) Sum of electronic and thermal Energies= -234.597023(Hartree/Particle) Sum of electronic and thermal Enthalpies= -234.597023(Hartree/Particle) Sum of electronic and thermal Free Energies= -234.595898(Hartree/Particle)
The Hart-tree optimisation of the "Chair" and "Boat" Transition Structures
Chair
This is the Hart-tree optimised anti2 conformer of 1,5-hexadiene file: opt_chair.

| Summary | Results |
|---|---|
| File name | chair_ts_ally_fragments_optimisation |
| File type | .log |
| Calculation Type | FREQ |
| Calculation Method | RHF |
| Basis set | 3-21G |
| Charge | 0 |
| Spin | Singlet |
| E(RHF) | -231.61932 a.u. |
| RMS Gradient Norm | 0.00005 a.u. |
| Imaginary Freq | |
| Dipole moment | 0.00 Debye |
| Point Group | C2h |
Optimising using the frozen coordinate method

| Summary | Results |
|---|---|
| File name | CHAIR_TS_ALLY_FRAGMENTS_OPTIMISATION_REDUNDANT |
| File type | .log |
| Calculation Type | FOPT |
| Calculation Method | RHF |
| Basis set | 3-21G |
| Charge | 0 |
| Spin | Singlet |
| E(RHF) | -231.61611 a.u. |
| RMS Gradient Norm | 0.00510 a.u. |
| Imaginary Freq | |
| Dipole moment | 0.01 Debye |
| Point Group | C2 |
Summary table for the Hessian optimised chair transition state

| Summary | Results |
|---|---|
| File name | CHAIR_TS_ALLY_FRAGMENTS_OPTIMISATION_hessian |
| File type | .log |
| Calculation Type | FOPT |
| Calculation Method | |
| Basis set | |
| Charge | 0 |
| Spin | Singlet |
| E | -231.50884 a.u. |
| RMS Gradient Norm | 0.02035 a.u. |
| Imaginary Freq | |
| Dipole moment | 0.28 Debye |
| Point Group | C1 |
Comparison between the "Hessian" optimised transition state and the "NoEigen" optimisation of the chair transition state.
The "Hessian" optimised transition state structure has an energy of -231.5088 a.u., this is higher in energy than the total energy of the "NoEigen" optimisation of the transition state (which has a total energy of -231.6913 a.u.). The NoEigen optimisation structure has a point group of C2h, while the Hessian optimised structure has a point group of C1. The bond forming/breaking lengths for the Hessian structure is 2.1587 and 2.2560 Å; the bond forming/breaking lengths for the NoEigen structure are both 2.0187 Å.
Boat

Summary table
| Summary | Results |
|---|---|
| File name | QST_optimised |
| File type | .log |
| Calculation Type | FREQ |
| Calculation Method | RHF |
| Basis set | 3-21G |
| Charge | 0 |
| Spin | Singlet |
| E(RHF) | -231.60280 a.u. |
| RMS Gradient Norm | 0.00011 a.u. |
| Imaginary Freq | 1 |
| Dipole moment | 0.16 Debye |
| Point Group | C2v |
This is the failed job file: opt_freq_failed_chair.
This is the successful job file: opt_freq_successful_chair.
Item table
Item Value Threshold Converged?
Maximum Force 0.000216 0.000450 YES
RMS Force 0.000052 0.000300 YES
Maximum Displacement 0.001770 0.001800 YES
RMS Displacement 0.000482 0.001200 YES
Predicted change in Energy=-6.861800D-07
Optimization completed.
-- Stationary point found.
Low frequencies
Low frequencies --- -839.1802 -9.2984 -0.0003 0.0007 0.0009 4.5924 Low frequencies --- 6.3001 154.7768 381.3735
QST3

The difference between the QST2 method and the QST3 method is that QST2 requires two molecule specifications, for the reactant and product, as its input, while QST3 requires three molecule specifications: the reactant, the product, and an initial structure for the transition state (in that exact order).
The QST3 optimised boat conformer has an imaginary vibrational mode at -840 cm-1; the QST2 optimised boat conformer, as mentioned previously, has an imaginary vibrational mode at -839 cm-1; the total energy of the QST3 optimised boat conformer is the same as that of the QST2 optimised boat conformer (-231.60280 a.u.).
Summary table
| Summary | Results |
|---|---|
| File name | QST3_optimised |
| File type | .log |
| Calculation Type | FREQ |
| Calculation Method | RHF |
| Basis set | 3-21G |
| Charge | 0 |
| Spin | Singlet |
| E(RHF) | -231.60280 a.u. |
| RMS Gradient Norm | 0.00002 a.u. |
| Imaginary Freq | 1 |
| Dipole moment | 0.16 Debye |
| Point Group | C2v |
QST3
Item table
Item Value Threshold Converged? Maximum Force 0.000069 0.000450 YES RMS Force 0.000013 0.000300 YES Maximum Displacement 0.001002 0.001800 YES RMS Displacement 0.000221 0.001200 YES Predicted change in Energy=-4.237594D-08 Optimization completed. -- Stationary point found.
Low frequencies
Low frequencies --- -839.8438 -5.7454 -4.0603 -0.0008 -0.0006 -0.0005 Low frequencies --- 0.5241 155.1918 382.0328
Intrinsic reaction coordinate (Chair)

The intrinsic reaction coordinate method was done on the "opt=redundant" optimized chair transition structure (using 50 points along the IRC). The last point on the IRC was optimised using the HF/3-21G level of theory. The last point on the IRC (before optimisation) has a total energy of -231.69158 a.u.; the optimised form of this structure has a total energy of 231.69167 a.u. - this corresponds to the gauche2 conformer.


Summary table
| Summary | Results |
|---|---|
| File name | optimisation_42_gauche2 |
| File type | .fch |
| Calculation Type | FOPT |
| Calculation Method | RHF |
| Basis set | 3-21G |
| Charge | 0 |
| Spin | Singlet |
| E(RHF) | -231.69167 a.u. |
| RMS Gradient Norm | 0.00000 a.u. |
| Imaginary Freq | 1 |
| Dipole moment | 0.38 Debye |
| Point Group | C2 |
This is the unsuccessfully optimised job file: IRC_chair.
This is the successfully optimised job file: 42_chair.
Optimisation of the chair and boat transitions structures using the B3LYP/6-31G* level of theory
Boat

The structure of the boat transition state is the same as what it was using the previous level of theory (point group is still C2v). The energy of the new transition state structure is -234.54309 a.u.; the structure is a transition state because the structure only has one imaginary vibrational mode at (-)530 cm-1.
The difference in total energy between the reactants (two Hart-tree optimised allyl fragments) and the boat Hart-tree optimised transition state is (-)0.04328 a.u.; the energy of the individual allyl fragments is -115.82304 a.u..
The difference in total energy between the reactants (two DFT optimised allyl fragments) and the boat DFT optimised transition state is (+)0.02239 a.u.; the energy of the individual allyl fragments is -117.26035 a.u..
Summary table
| Summary | Results |
|---|---|
| File name | BOAT_B3LYP6-31G_OPTIMISATION |
| File type | .log |
| Calculation Type | FTS |
| Calculation Method | RB3LYP |
| Basis set | 6-31G(d) |
| Charge | 0 |
| Spin | Singlet |
| E(RB3LYP) | -234.54309 a.u. |
| RMS Gradient Norm | 0.00000 a.u. |
| Imaginary Freq | 1 |
| Dipole moment | 0.06 Debye |
| Point Group | C2v |
Item table
Item Value Threshold Converged?
Maximum Force 0.000009 0.000450 YES
RMS Force 0.000003 0.000300 YES
Maximum Displacement 0.000141 0.001800 YES
RMS Displacement 0.000045 0.001200 YES
Predicted change in Energy=-2.565747D-09
Optimization completed.
-- Stationary point found.
Low frequencies
Low frequencies --- -530.3604 -8.4039 -0.0002 0.0008 0.0012 15.4620 Low frequencies --- 17.6139 135.6118 261.6999
Summary of energies
| B3LYP/6-31G* | |||||||
|---|---|---|---|---|---|---|---|
| Electronic energy (a.u.) | zero-point energy (a.u.) | Sum of electronic and zero-point energies (a.u.) | Sum of electronic and zero-point energies (KCalmol-1)
|
Thermal energy (a.u.) | Sum of electronic and thermal energies (a.u.) | Sum of electronic and thermal energies (KCalmol-1) | |
| at 0 K | at 0 K | at 0 K | at 298.15 K | at 298.15 K | at 298.15 K | ||
| Boat TS | -234.54309 | 0.12567 | -234.41742 | -1.47099 x105 | 0.13265 | -234.41044 | -1.47095 x105 |
| Reactant (anti2) | -234.61171 | 0.12724 | -234.48447 | -1.47141 x105 | 0.13517 | -234.47654 | -1.47136 x105
|
The activation energy at 0 K is the difference between the "sum of electronic and zero-point energies" for the boat transition state and the anti2 conformer. From the tabulated results above, the activation energy is 42 KCalmol-1. Similarly, the activation energy at 298.15 K is the difference between the "sum of electronic and thermal energies" for the boat transition state and the anti2 conformer. From the tabulated results above, the activation energy is 41 KCalmol-1. The activation energy calculated at 0 K gives us a result that is slightly closer to the experimental value of 44.7 ± 2.0 KCalmol-1 (at 0 K).
Chair
The structure of the chair transition structure is the same as what it was using the previous level of theory (point group is still C2h). The energy of the new transition state structure is -234.55698 a.u.; this structure is a transition state because it only has one imaginary vibrational mode at (-)566 cm-1.
The difference in total energy between the reactants (two Hart-tree optimised allyl fragments) and the chair Hart-tree optimised transition state is (+)0.02676 a.u.; the energy of the individual allyl fragments is -115.82304 a.u..
The difference in total energy between the reactants (two DFT optimised allyl fragments) and the chair DFT optimised transition state is (+)0.03628 a.u.; the energy of the individual allyl fragments is -117.26035 a.u..

Summary table
| Summary | Results |
|---|---|
| File name | CHAIR_TS_ALLYL_631GD |
| File type | .log |
| Calculation Type | FTS |
| Calculation Method | RB3LYP |
| Basis set | 6-31G(d) |
| Charge | 0 |
| Spin | Singlet |
| E(RB3LYP) | -234.55698 a.u. |
| RMS Gradient Norm | 0.00003 a.u. |
| Imaginary Freq | 1 |
| Dipole moment | 0.00 Debye |
| Point Group | C2h |
opt_chair and freq_chair.
Item table
Item Value Threshold Converged?
Maximum Force 0.000053 0.000450 YES
RMS Force 0.000011 0.000300 YES
Maximum Displacement 0.000988 0.001800 YES
RMS Displacement 0.000246 0.001200 YES
Predicted change in Energy=-5.051083D-08
Optimization completed.
-- Stationary point found.
Low frequencies
Low frequencies --- -18.3528 -14.6266 -11.5337 0.0006 0.0008 0.0009 Low frequencies --- 63.3097 97.1475 103.5644
Summary of energies
| B3LYP/6-31G* | |||||||
|---|---|---|---|---|---|---|---|
| Electronic energy (a.u.) | zero-point energy (a.u.) | Sum of electronic and zero-point energies (a.u.) | Sum of electronic and zero-point energies (KCalmol-1)
|
Thermal energy (a.u.) | Sum of electronic and thermal energies (a.u.) | Sum of electronic and thermal energies (KCalmol-1) | |
| at 0 K | at 0 K | at 0 K | at 298.15 K | at 298.15 K | at 298.15 K | ||
| Chair TS | -234.55698 | 0.12683 | -234.43015 | -1.47107 x105 | 0.13338 | -234.42360 | -1.47103 x105 |
| Reactant (anti2) | -234.61171 | 0.12724 | -234.48447 | -1.47141 x105 | 0.13517 | -234.47654 | -1.47136 x105
|
The activation energy at 0 K is the difference between the "sum of electronic and zero-point energies" for the chair transition state and the anti2 conformer. From the tabulated results above, the activation energy is 34 KCalmol-1. Similarly, the activation energy at 298.15 K is the difference between the "sum of electronic and thermal energies" for the chair transition state and the anti2 conformer. From the tabulated results above, the activation energy is 33 KCalmol-1. The activation energies that were calculated at 0 K and 298.15 K are both equally as close to the experimental value of 33.5 ± 0.5 KCalmol-1 (at 0 K) - and they are both within the region of uncertainty.
Final comments about the tutorial
No problems were encountered whilst undertaking this tutorial.
The Diels Alder Cycloaddition
1,3-Butadiene

HOMO and LUMO
| Molecular Orbital | Diagram | Bonding or Antibonding? | Energy (a.u.) | Symmetry |
|---|---|---|---|---|
| HOMO |  |
Bonding. | -0.35322. | Symmetric. |
| LUMO |  |
Antibonding. | 0.01136. | Antiymmetric. |
Summary table
| Summary | Results |
|---|---|
| File name | BUTADIENE_OPTIMISATION |
| File type | .log |
| Calculation Type | FOPT |
| Calculation Method | RPDDG |
| Basis set | ZDO |
| Charge | 0 |
| Spin | Singlet |
| E(RPDDG) | 0.04591539 a.u. |
| RMS Gradient Norm | 0.00003 a.u. |
| Imaginary Freq | |
| Dipole moment | 0.06 Debye |
| Point Group | C2 |
Cyclohexene
HOMO and LUMO of the transition state
| Molecular Orbital | Diagram | Bonding or Antibonding? | Energy (a.u.) | Symmetry |
|---|---|---|---|---|
| HOMO |  |
Bonding. | -0.21896. | Symmetric. |
| LUMO |  |
Antibonding. | -0.00861. | Symmetric. |
Structure

| Summary | Results |
|---|---|
| File name | CYCLOHEXENE_OPTIMISATION_frequency |
| File type | .fch |
| Calculation Type | FOPT |
| Calculation Method | RB3LYP |
| Basis set | 6-31G(d) |
| Charge | 0 |
| Spin | Singlet |
| E(RB3LYP) | -234.54390 a.u. |
| RMS Gradient Norm | 0.00002 a.u. |
| Imaginary Freq | 1 |
| Dipole moment | 0.39 Debye |
| Point Group |
Bond lengths
| Bond | Bond length (Å) | Hybridisation |
|---|---|---|
| C4-H6 | 1.08909. | sp2 - H |
| C9-H11 | 1.08420. | sp3 - H |
| C9-H10 | 1.08628. | sp3 - H |
| C12-H11 | 1.08418. | sp3 - H |
| C12-H13 | 1.08628. | sp3 - H |
| C1-H5 | 1.08907. | sp2 - H |
| C3-H7 | 1.08731. | sp2.5 - H |
| C3-H15 | 1.08404.. | sp2.5 - H |
| C2-H8 | 1.08732. | sp2.5 - H |
| C2-H16 | 1.08404. | sp2.5 - H |
| C4=C1. | 1.40722. | sp2 - sp2 |
| C9-C12. | 1.38598. | sp3 - sp3 |
| C4-C3. | 1.38307. | sp2 - sp2.5 |
| C1-C2. | 1.38313. | sp2 - sp2.5 |
| C9-------C3. | 2.27207 | sp3 - sp2.5 |
| C12-------C2. | 2.27235 | sp3 - sp2.5 |
The typical sp3 - sp3 C-C bond length is 1.54 Å; the typical sp3 - sp2 C-C bond length is 1.50 Å; and the typical sp2 - sp2 C-C bond length is 1.47 Å.[1] The Van der Waals radius of a C atom is 1.70 Å.[2] A covalent radius (twice the distance of a covalent bond) is generally smaller than the Van der Waals radius because the Van der waals radius is double the distance of closest approach between two non-bonded atoms (of the same element) that are merely colliding with one another without reacting to form a bond. This is the reason why the bond length of the partly formed σ C-C bonds in the transition state of cyclohexene is smaller than 3.4 Å.
Vibration
The formation of the two new bonds is synchronous. The vibration that corresponds to the reaction path at the transition state is a stretching vibrational mode, whereas the lowest positive frequency is a twisting vibrational mode; ethylene and s-cis butadiene vibrate as if they are already a single molecule in the lowest positive vibrational mode, whereas ethylene and s-cis butadiene vibrate as if they are separate molecules in the vibrational modes that corresponds to the reaction path at the transition state.


Low frequencies
Low frequencies --- -525.1800 -5.3446 -0.0007 -0.0006 -0.0004 10.9440 Low frequencies --- 20.0558 135.8245 203.7598
Item table
Item Value Threshold Converged? Maximum Force 0.000057 0.000450 YES RMS Force 0.000009 0.000300 YES Maximum Displacement 0.001795 0.001800 YES RMS Displacement 0.000504 0.001200 YES Predicted change in Energy=-4.090426D-08 Optimization completed. -- Stationary point found.
MO diagram



The reaction is allowed because, as illustrated on the MO diagram above, the HOMO of ethylene can interact with the LUMO of 1,3-Butadiene and the HOMO of 1,3-Butadiene can interact with the LUMO ethylene. Also, the electrons of the cyclohexene transition state are only occupying bonding (or non-bonding) MOs; the transition state is stabilised because no electron is occupying an antibonding MO.
Stereoselectivity of the Diels Alder Reaction
The exo Diels-Alder transition state adduct has a total energy of -612.67931 a.u., whileas the endo Diels-Alder transition state adduct has a total energy of -612.68340 a.u. - this means that the endo Diels-Alder product is the kinetic product. The C=O bonds of maleic anhydride are pointing in the direction of cyclo-1,3-hexadiene's pi-system in the endo transtion state, whileas the C=O bonds of maleic anhydride are pointing away from cyclo-1,3-hexadiene's pi-system in the exo transition state.
The exo transition state is less sterically strained because the substituents from maleic anhydride (the dienophile) is pointing towards the smaller bridge (and is pointing away from the larger bridge); the reason why the endo transitiion state is more sterically strained is because the substituents from the dienophile is pointing towards the larger bridge (and is pointing away from the smaller bridge).
The structures that are shown on the left are the transition structures for the Diels-Alder reaction because both structures only have one imaginary frequency mode.
HOMO and LUMO of the endo transition state
| Molecular Orbital | Diagram | Energy (a.u.) | Symmetry |
|---|---|---|---|
| HOMO |  |
-0.24230. | Antisymmetric. |
| LUMO |  |
-0.06773. | Antisymmetric. |
Summary table
| Summary | Results |
|---|---|
| File name | ENDO_DIELS_ALDER |
| File type | .log |
| Calculation Type | FREQ |
| Calculation Method | RB3LYP |
| Basis set | 6-31G(d) |
| Charge | 0 |
| Spin | Singlet |
| E(RB3LYP) | -612.68340 a.u. |
| RMS Gradient Norm | 0.000008 a.u. |
| Imaginary Freq | 1 |
| Dipole moment | 6.11 Debye |
| Point Group | C1 |
Item table
Item Value Threshold Converged?
Maximum Force 0.000023 0.000450 YES
RMS Force 0.000003 0.000300 YES
Maximum Displacement 0.000455 0.001800 YES
RMS Displacement 0.000122 0.001200 YES
Predicted change in Energy=-5.350598D-09
Optimization completed.
-- Stationary point found.
Low frequencies
Low frequencies --- -447.0370 -14.2013 -0.0007 0.0003 0.0004 4.6897 Low frequencies --- 11.2730 59.6773 118.3724
HOMO and LUMO of the exo transition state
| Molecular Orbital | Diagram | Energy (a.u.) | Symmetry |
|---|---|---|---|
| HOMO |  |
-0.24215. | Antisymmetric. |
| LUMO |  |
-0.07842. | Antisymmetric. |
Summary table
| Summary | Results |
|---|---|
| File name | EXO_DIELS_ALDER |
| File type | .log |
| Calculation Type | FREQ |
| Calculation Method | RB3LYP |
| Basis set | 6-31G(d) |
| Charge | 0 |
| Spin | Singlet |
| E(RB3LYP) | -612.679311 a.u. |
| RMS Gradient Norm | 0.00002 a.u. |
| Imaginary Freq | 1 |
| Dipole moment | 5.55 Debye |
| Point Group | C1 |
Item table
Item Value Threshold Converged?
Maximum Force 0.000076 0.000450 YES
RMS Force 0.000009 0.000300 YES
Maximum Displacement 0.001334 0.001800 YES
RMS Displacement 0.000367 0.001200 YES
Predicted change in Energy=-1.689065D-08
Optimization completed.
-- Stationary point found
Low frequencies
Low frequencies --- -448.2368 -13.9880 -11.9194 0.0002 0.0008 0.0010 Low frequencies --- 3.1396 53.2571 109.0993
Bond lengths
Bond lengths for the exo adduct
| Bond | Bond length (Å) | Hybridisation |
|---|---|---|
| C3-C8 | 1.51466. | sp2.5 -
sp3 |
| C8-C11 | 1.55838. | sp3 -
sp2 |
| C11-C2 | 1.51451. | sp3 -
sp2.5 |
| C19-C15 | 1.47939. | sp3 -
sp2 |
| C18-C17 | 1.47945. | sp3 -
sp2 |
| C1===C4 | 1.40352. | sp2 -
sp2 |
| C4===C3 | 1.39138. | sp2 -
sp2.5 |
| C1===C2 | 1.39133. | sp2 -
sp2.5 |
| C19===C18 | 1.39785. | sp2.5 -
sp2.5 |
| C19-----C3 | 2.29117. | sp2.5 -
sp2.5 |
| C18-----C2 | 2.29056. | sp2.5 -
sp2.5
|
The C-C through space distances between the -(C=O)-O-(C=O)- fragment of the maleic anhydride and the C atoms of the “opposite” -CH2-CH2- for the exo transition state is between 3.02801 (C17 and C11) - 3.02813 (C15-C8) Å.
Bond lengths for the endo adduct
| Bond | Bond length (Å) | Hybridisation |
|---|---|---|
| C3-C8 | 1.51511. | sp2.5 -
sp3 |
| C8-C11 | 1.55824. | sp3 -
sp2 |
| C11-C2 | 1.51507. | sp3 -
sp2.5 |
| C19-C15 | 1.47948. | sp3 -
sp2 |
| C18-C17 | 1.47951. | sp3 -
sp2 |
| C1===C4 | 1.40307. | sp2 -
sp2 |
| C4===C3 | 1.39125. | sp2 -
sp2.5 |
| C1===C2 | 1.39124. | sp2 -
sp2.5 |
| C19===C18 | 1.39398. | sp2.5 -
sp2.5 |
| C19-----C2 | 2.26837. | sp2.5 -
sp2.5 |
| C18-----C3 | 2.26829. | sp2.5 -
sp2.5
|
The C-C through space distances between the -(C=O)-O-(C=O)- fragment of the maleic anhydride and the “opposite” -CH=CH- fragment for the endo transition state is 2.99007 Å.
Secondary orbital interactions

The exo transition state is the thermodynamically favoured transition state because, as mentioned earlier, the exo transitiion state is less sterically strained. The endo transition state is the kinetically favoured transition state because of the so called “secondary orbital overlap effect”. As mentioned previously, the C=O bonds of maleic anhydride are pointing in the direction of cyclo-1,3-hexadiene's pi-system in the endo transtion state. The HOMO of cyclo-1,3-hexadiene is able to overlap with the LUMO of maleic anhydride - this lowers the energy of the transition state relative to that of the exo transition state; this interaction is not present in the exo transition state because the C=O bonds of maleic anhydride are pointing away from cyclo-1,3-hexadiene's pi-system.
The HOMO of the endo transition state has four strong σ bonding interactions between the p orbitals of the -(C=O)-O-(C=O)- fragment and the p orbitals from cyclo-1,3-hexadiene; the HOMO has four nodes in total. The HOMO of the exo transition state, on the other hand, has four strong σ antibonding interactions between the p orbitals of the -(C=O)-O-(C=O)- fragment and the p orbitals from cyclo-1,3-hexadiene; the HOMO has eight nodes in total.
In conclusion, the secondary orbital overlap effect helps to stabilise the endo transition state, this is not the case for the exo transition state; the strong σ antibonding interactions destabilise the exo transition state, relative to the endo transition state.
Further comments
Electronic effects (the secondary overlap orbital effect), steric effects, stereospecificity effects (the idea that the stereochemistry of the dienophile is always maintained because the diene undergoes a syn addition to the dienophile), and stereoselectivity (the endo being the favoured product) were all included in the calculations of the Diels-Alder transition states. The effect of electron donating groups on the diene facilitate reaction (regioselectivity) was neglected because both ethylene doesn't have any electron withdrawing substituents - such as a carbonyl group, an amine group etc., and maleic anhydride has two carbonyl groups, but the carbonyl groups are both the same distance away from the double bond (maleic anhydride is symmetric, with respect to the double dond). Also, 1,3-butadiene and cyclo-1,3-hexadiene don't have any electron releasing substituents. The regioselectivity effect is based upon the idea that the carbon (of the dienophile) that bears the most electron withdrawing substituent will bond to the carbon (of the diene) that bears the most electron releasing substituent, and vise versa.
The question of whether Diels-Alder reactions using a more electron rich version of the same diene can give lower or higher activation energies (and how do these transition structures vary, with respect to each other), when reacted with the same dienophile, was neglected. Similarly, The question of whether Diels-Alder reactions using a more electron poor version of the same dienophile can give lower or higher activation energies (and how do these transition structures vary, with respect to each other), when reacted with the same diene, was also neglected.
In conclusion, the results that were derived from this experiment can be explained using various theories within physical and organic chemistry.
References
- ↑ http://pubs.rsc.org/en/content/articlepdf/1987/p2/p298700000s1 F.H. Allen et al., Tables of bond lengths determined by X-ray and neutron diffraction. Part 1. Bond lengths in organic compounds, J. CHEM. SOC, 1987, 1-19.
- ↑ Rowland RS, Taylor R (1996). "Intermolecular nonbonded contact distances in organic crystal structures: comparison with distances expected from van der Waals radii". J. Phys. Chem. 100 (18): 7384–7391. doi:10.1021/jp953141
- ↑ Ian Fleming (1976), "Frontier Orbitals and Organic Chemical Reactions", Wiley-Blackwell, 120 - 160.