
Organic:pericyclic historical
Historical Background.
Prior to the 1960s, organic reactivity was thought to be dominated by factors which included:
- the relative stability of reactant and product (i.e. thermodynamic control),
- geometrical effects such as strain, steric interactions, hydrogen bonding, neighbouring group effects (entropy),
- electrostatic effects such as the polarity of functional groups (eg the carbonyl group) and
- the aromaticity of either the reactant or the product.
During the course of the synthesis of vitamin B12 in the early 1960s, Robert Woodward concluded that none of the above factors could rationalise the following reaction. It was thought at the time using simple molecular models (the quantitative aspect of which was pioneered in 1951 by another great organic chemist and Nobel prize winner, Derek Barton) that the actual product would in fact be less stable (and hence less likely to form) than the other isomer.

A new explanation was developed based on 'stereoelectronic' factors, i.e. recognising that the three-dimensional properties of the electrons and their phase relationship could dominate the other factors listed above.
This theory of stereoelectronic control of pericyclic reactions was derived using an approach known as the conservation of orbital symmetry, and together with the theoretician Roald Hoffmann, Woodward was eventually awarded the Nobel prize for this work (DOI: | J. Am. Chem. Soc., 1965, 87, 395 and succeeding articles ). The recognition that stereoelectronic factors (more accurately the three dimensional properties of the molecular wavefunction) can be critical to explaining reactivity has had an enormous impact on understanding organic chemistry (see Introduction to stereoelectronics).
Definition of Pericyclic Reactions.
A pericyclic reaction is one in which bonds are made or broken in a concerted cyclic transition state. A concerted reaction is one which involves no intermediates during the course of the reaction. A stepwise and therefore non-concerted and non-pericyclic reaction is shown on the right.

Pericyclic reactions have certain characteristic properties, although as usual it is not difficult to find exceptions to all these rules.
- There is a relatively small solvent effect on the rate of reaction (unless the reactants themselves happen to be charged, ie carbonium or carbanions). Pericyclic reactions can occur in the gas phase with no solvent. Quite recently, the use of water to accelerate pericyclic reactions (by perhaps a factor of 10 to 100) has been much investigated, but the acceleration is largely due to the formation of hydrogen bonds specific to the transition state.
- There is no nucleophilic or electrophilic component. This means that in the arrow pushing sense, there is no beginning and no ending for the arrows, and the arrow pushing can occur in either a clockwise or anti-clockwise direction.
- Normally, no catalyst is need to promote the reactions. However, many transition metal complexes can catalyse pericyclic reactions by virtue of d-orbital participation. Lewis acids also catalyse many forms of pericyclic reactions, either directly, or by changing the mechanism of the the reaction so that it becomes a stepwise process (ie the right hand diagram above) and hence no longer a true pericyclic reaction. It is also possible to accelerate the reactions by the use of pressure for those reactions involving a substantial decrease in volume, and catalysts ("[inclusion_phenomena.pdf molecular containers]") also exist which provide suitable [#DAase "cavities"] for promoting pericyclic reactions such as cycloadditions.
- Pericyclic reactions normally show a very high stereospecificity.
- Pericyclic reactions can be frequently promoted by light (denoted hν in the text) as well as heat (denoted Δ in the text). Normally, the stereochemistry under the two sets of conditions is different, and it was (originally) thought invariably opposite. Current thinking about the photochemical route is more complex!
- Unlike nucleophilic/electrophilic reactions, pericyclic reactions are unusual in that few enzymes which catalyse them are known. Artificial catalytic antibodies ('abzymes') can be created which can perform this feat, [#DAase "Diels-Alderase"] does this for cycloadditions and [#chorismate Chorismate mutase] for the Claisen rearrangement.
Principal Categories of Organic Pericyclic Reactions.
Here we make no attempt to describe the rich variety of inorganic reactivity that can also be described as pericyclic. There are four major organic categories covering a very wide scope of organic and organometallic chemistry, and which form a rich basis for understanding the more subtle aspects of synthesis and organic chemistry.
Electrocyclic Reactions.
An electrocyclic reaction involves the concerted formation of a σ bond between the two ends of a linear conjugated π system, or the reverse reaction in which the σ bond is broken to produce a linear conjugated system.  In these definitions, by 'conjugated', we mean a system of C-C multiple bonds, or of lone pairs. Also;
In these definitions, by 'conjugated', we mean a system of C-C multiple bonds, or of lone pairs. Also; 
Cycloaddition and Cyclo-elimination reactions
A cycloaddition reaction involves the concerted formation of two or more σ bonds between the termini of two or more conjugated π systems. The reverse reaction involves the concerted cleavage of two or more σ bonds to produced two or more conjugated π systems.  For the specific case where formation or cleavage of two σ bonds occurs at a single atom, the reaction is called a Cheletropic reaction.
For the specific case where formation or cleavage of two σ bonds occurs at a single atom, the reaction is called a Cheletropic reaction. 
Sigmatropic Reactions
A sigmatropic reaction involves the concerted migration of an atom or group from one point of attachment to a conjugated system to another point of attachment, during which one σ bond is broken and another σ bond is made. A sigmatropic reaction can be classified according to the length of the group that migrates, and the length of the backbone along which it migrates; 
Ene Reactions
A general category which involves the formation and cleavage of unequal numbers of σ bonds in a concerted cyclic transition state. 
Another "in-between" category is those involving cyclopropane rings, which can also act as alkenes, and as such participate in pericyclic reactivity. 
Should this be regarded as closer to an electrocyclic reaction or a sigmatropic shift?
Metallo and other Synthetically Useful Reactions
Many diverse reactions of synthetic interest could be classified as pericyclic. For example, Chromium(VI) (and other metal) oxidations of alcohols proceed via cyclic transition states. Depending on whether chromate or dichromate is used, this would be either a 6 or an 8 electron process.  Selenium dioxide oxidations and Al-mediated hydride reductions show similar properties. Other elements also show pericyclic mechanisms, a good example of which involves boron (X here is a chiral auxilliary);
Selenium dioxide oxidations and Al-mediated hydride reductions show similar properties. Other elements also show pericyclic mechanisms, a good example of which involves boron (X here is a chiral auxilliary); 
Co-arctic Reactions
A new category which involves the formation or cleavage of four σ bonds at a single atomic centre.  In this category also rank Br+ and oxygen atom transfer reactions between alkenes.
In this category also rank Br+ and oxygen atom transfer reactions between alkenes.
Pseudopericyclic Reactions
In a pseudopericyclic reaction, there is no continuous orbital overlap around the ring of breaking and forming bonds. All pseudo-pericyclic reactions are allowed; there are no anti-aromatic transition states. Pseudo-pericyclic reactions can have lower barriers than pericyclic alternatives.
Theoretical Explanation.
The original explanation of Woodward and Hoffmann was formalised by Longuet-Higgins and Abrahamson (DOI: 10.1021/ja01087a033, see also 10.1021/ja01087a033), and involved generating a so called "orbital "correlation diagram" for the reaction under consideration, and then carrying out the reaction in such a manner that the symmetries of the reactant and product orbitals matched exactly. Such an approach, whilst theoretically rigorous, is not readily applicable to the majority of more complex reactions. Two much simpler methods have been outlined, the first of which will be expanded in more detail, whilst the second will only be described briefly;
Conservation of Orbital Symmetry (Longuet-Higgins and Abrahamson)
Let us first define the symmetry properties of a 1s and a 2p orbital with respect to a plane of symmetry or an axis of symmetry as shown below;
 One can take this one step further by considering the symmetry properties of molecular orbitals formed by the overlap of two or more atomic orbitals;
One can take this one step further by considering the symmetry properties of molecular orbitals formed by the overlap of two or more atomic orbitals;
- MOs formed from two overlapping σ orbitals:
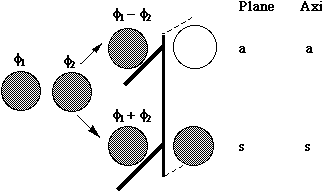
- MOs formed from two overlapping p orbitals (σ bonds):
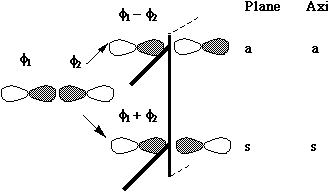
- MOs formed from two parallel overlapping p orbitals (π bonds):




We can now use these basic orbitals to construct the relevant molecular orbitals for two interconverting molecules, cyclobutene and butadiene, with the purpose of following how these two sets of orbitals change when one molecule is converted into the other. Note particularly that we need only construct the MOs explicitly involved in the reaction; most of the σ framework remains unchanged and no orbitals derived from this need to be considered:
|
|
|
|
|
|










In order to interconvert cyclobutene and butadiene, the four MOs labelled ψ1, ψ2, ψ3, ψ4 must be converted into ψσ, ψπ, ψπ*, ψσ*. There are two stereochemically distinct ways in which this might be accomplished; conrotation;
disrotation;

This enables a correlation diagram for the reaction to be constructed, according to the following rules: no two orbitals of the same symmetry can cross during the reaction, whilst orbitals of different symmetry can cross. The favoured pathway is the one which results in a product of the same electronic excitation as the reactant. A pathway which results in the product in a higher electronic state than the reactant is said to be "forbidden".

Whilst this rule is normally followed fairly well for ground states, it can be overturned when for example steric or geometrical strain in the "allowed" pathway promotes the "forbidden" route. The situation is actually more complex for photochemical reactions, and much recent evidence suggests that the Woodward-Hoffmann rules are not always followed. These correlation diagrams can be generalised for any electrocyclic reaction with appropriate symmetry. However, correlation diagrams are less readily applied for reactions with no symmetry. Dewar and Zimmerman independently noticed that the 'topological' properties of these correlation diagrams are very similar to those obtained using eg Huckel theory for aromatic molecules. For example, the diagram for the electrocyclic conversion of hexatriene to cyclohexadiene is remarkably similar at the transition state to the ground state orbitals of benzene;

Alternative Approach: Transition State Aromaticity (Dewar and Zimmermann)
Huckel Aromatics
The suprafacial mode orbital correlation diagram for hexatriene can be generalised by reference to benzene. The benzene molecule has the same 'suprafacial' topology, by which we mean that the π electron density in benzene is continuous along the top or bottom face of the molecule. If the transition state for the pericyclic reaction has the same topology, it is said to resemble "Hückel" topology, after Erich Hückel who first indicated why a molecule such as benzene should be especially stable.
 [#BEN
[#BEN  ]
]
Thus a 'suprafacial' or 'Hückel' transition state in a pericyclic reaction is associated with a plane of symmetry and is particularly favourable if the number of cyclically conjugated π electrons in the transition state equals 4n+2 (the Huckel rule, where n = 0, 1, 2 etc).
Huckel was also able to show that if a cyclic conjugated π system is irradiated with light so that it goes into the first excited singlet or triplet electronic state it is especially stable if the number of cyclically conjugated electrons equals 4n. Hence photochemically activated pericyclic reactions will proceed suprafacially via a Huckel transition state if the electron count corresponds to 4n.
Möbius Aromatics (Heilbronner)
The antarafacial mode described above is said to resemble "Möbius" topology, after August Ferdinand Möbius, who invented the famous strips (or possibly it was Johann Benedict Listing). An antarafacial mode can be formed by taking a cyclic alkene "strip" and giving the π system a 180° (half) twist (and in doing so for a molecule a two-fold axis of symmetry is created in the resulting object). [c8f8mm.3dmf
[c8f8mm.3dmf  ]
]
Edgar Heilbronner in 1964 worked out that such a twisted system would be stable (latter shown to be aromatic) if it contained 4n conjugated π electrons. No actual examples were identified until relatively recently, when it was proposed that [12], [16] and [20] annulenes have several [#12 higher energy conformations] which adopt this mode. In 2003, the first crystal structure of a true stable Möbius [16] annulene was completed, and various heteroannulenes were identified as existing in Möbius form (DOI: 10.1021/cr030092l
Just as with excited state Hückel aromatics, Möbius molecules in the excited singlet or triplet state are thought to be aromatic if they contain 4n+2 rather than 4n π electrons. The example to the right shows C8F82- (perfluorocyclo-octatetraene dianion, a 4n+2 electron system) which as a triplet is Möbius-aromatic (these examples were all discovered at Imperial College, several as part of 4th year student projects).
These four types of aromaticity are most easily summarised as follows;
The Pericyclic Reaction Selection Rules
- A thermally activated pericyclic reaction will proceed via a Huckel topology containing only suprafacial components if the cyclically conjugated π electrons equal 4n+2 (n=0,1,2 etc).
- A photochemically activated pericyclic reaction may proceed via a Huckel topology containing only suprafacial components if the cyclically conjugated π electrons equal 4n.
- A thermally activated pericyclic reaction will proceed via a Möbius topology containing ONE antarafacial component if the cyclically conjugated π electrons equal 4n (n=0,1,2 etc).
- A photochemically activated pericyclic reaction may proceed via a Möbius topology containing ONE antarafacial component if the cyclically conjugated π electrons equal 4n+2 (n=0,1,2 etc).
A useful mnemonic for remembering these rules is due to Frost, Musulin and Zimmermann:

2005: Higher-twist Möbius Rings
In 2005, the Möbius approach has been extended to rings created by imparting more than just one half-twist to a cylinder. Such systems, like the single half-twist topology, are also chiral and can have a C2 axis of symmetry (i.e. the two twists do not cancel). DOI: 10.1039/b510508k and 10.1021/ol0518333
Alternative Approach: The Frontier Orbital Method (Fukui)
(The following is optional and is intended for use in tutorials). This involves using the principles of quantum mechanics to generate the Highest Occupied Molecular Orbital (HOMO) and the Lowest Unoccupied Molecular Orbital (LUMO) for the reactant(s). The formation of the new &σ; bond must occur by appropriate overlap of the nodes of these orbitals. If only one &σ; bond is forming, as in an electrocyclic reaction, then only the overlap of the HOMO of the reactant is considered. Such overlap can occur in one of two fundamental ways; The suprafacial mode involves each component of the new σ bond being formed from the SAME face of the reactant π system. See DOI: 10.1016/S0040-4039(01)83901-0.
 [hexatriene.3dmf
[hexatriene.3dmf  ]
]
The antarafacial mode involves a 'twisting' of the orbitals so that the two components of the new &σ; bond come from OPPOSITE faces of the reactant π system. If two or more &σ; bonds form during the reaction, as in cycloaddition reactions, then the overlap of the HOMO of one reactant with the LUMO of the second reactant must be considered. For simple systems, the form of the HOMO and LUMO is not difficult to remember. For more complex systems, explicit calculations have to be carried out and the Frontier Orbital method becomes more difficult to apply. The advantage of the "FMO" method is that it can be expressed quantitatively in terms of the magnitude of the coefficients involved, and can hence be used to predict regioselectivity etc.
Modern Approach: The Transition State Potential Energy Surface Method
The next level of theory beyond the FMO method involves the explicit location of the pericyclic transition state using a Quantum Mechanical method. A complete worked example of such modelling of a pericylic reaction can be seen here (and is dealt with in detail in another lecture course).
Examples of Pericyclic Reactions
Worked examples can be seen in the next section of these notes.
--Rzepa 19:15, 12 April 2006 (bst)