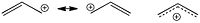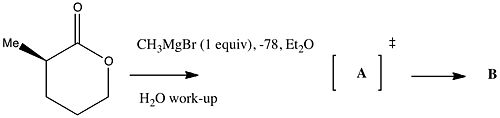Most organic (synthetic) chemists tend to follow a set of conventions in drawing out their reactions. One quickly learns that these are not necessarily (IUPAC) standards, but part of the commonly accepted folklore. As such, it can be sometimes difficult to find out what these mean in more formal textbooks. Sometimes even lecturers and tutors forget that they have to be learnt, and just assume they are absorbed by osmosis or other process. So collected here are some of the conventions that might have otherwise slipped through the nets of lectures and tutorials.
Timescales in chemistry
Attosecond, 10-18 s. Electron dynamics. The time taken for an electron to traverse the 1s orbit in a hydrogen atom.
Femtosecond, 10-15 s. Nuclear vibrations. Time taken for a normal vibration, of eg a C-H bond (e.g set ΔG‡ to zero in the equation Ln(k/T) = 23.76 - ΔG‡/RT)
Picosecond, 10-12 s.
Nanosecond, 10-9 s. Time taken for a diffusion controlled reaction, such as proton transfer.
Microsecond, 10-6 s.
Millisecond, 10-3 s. Time taken for the relaxation process of a nucleus precessing in a magnetic field, the NMR timescale.
Second, 100 s.
Kilosecond, 103 s. The normal time for a synthetic reaction in a laboratory. Boundary between a conformation and a configuration.
Megasecond, 106 s. Normal limit for a synthetic reaction (reflux etc).
Gigasecond, 109 s.
Terasecond, 1012 s.
Petasecond, 1015 s.
Exasecond, 1018 s. Age of universe. Less than the time taken for a molecule of alanine to racemise.
See Take a number: DOI:10.1038/nchem.1733 on "anchoring values' in chemistry, very much an under-rated skill perhaps?
The molecular formula
The formula by convention is set out as follows: carbon count first, then hydrogen, then the elements in alphabetic order, as C2H2BFLiNO
If the compound consists of two separate components, unconnected by any bond, then two formulae for each component are displayed, with a dot between them, as NH4+.NO2-
The formula can be used to compute double bond equivalents. A core set of rules for doing so is set out here
Abbreviations
Whilst a molecular formula uses only elemental symbols to represent the constitution of a molecule, it is also convenient to have a shorthand notation for groups of atoms in a molecule. The use of such abbreviations tends to be relatively informal, but there are some implicit rules;
Use an abbreviation only when you do not want to involve the bonds within group in a reaction mechanism, i.e. it is common to both reactant and product.
Use an abbreviation as a wild-card which can have several different values, which may or may not be listed.
Examples
The examples below are by no means comprehensive. Add your own favourites!
Alkyl groups: Me (CH3), Et (C2H5), n-Pr (1-propyl), i-Pr (2-propyl), Bu (1-butyl), t-Bu (2-methylpropyl). Pentyl and above are not normally abbreviated.
Groups with phenyl rings: Ph (Phenyl, C6H5), Bz (phenylmethane), p-Tol (p-methylphenyl).
Carbonyl systems: Ac (acetyl, CH3CO).
Variable groups: R (normally means any alkyl group), Ar (normally means any aromatic group), X (can mean almost anything).
Leaving groups. Electronegative groups that can accept electrons and leave in a reaction: Ts (p-methylphenylsulfonate), Ms (methylsulfonate), Tf (trifluoromethylsulfonate).
Metal ligands. acac (acetylacetonate, an old fashioned name)
etc etc etc
The (covalent) bond
Lewis in 1916 (DOI:10.1021/ja02261a002 ) proposed the electron-pair covalent bond, in which electrons are shared (~equally) between two atoms.
We now take a single (by convention straight, although that is in fact an approximation in some molecules) line drawn between two atoms to indicate such a Lewis single bond sharing two electrons.
One exception to the use of straight lines is sometimes illustrated by cyclopropane, where the lines connecting the carbons can be drawn as curved.
Lewis also recognised the shared four-electron bond, which we now indicate with two lines drawn between a pair of atoms as a double bond.
Although the two drawn lines appear identical, they imply quite different origins (σ and π in this case, see below).
If you want to see a classical historical error due to not distinguishing between the σ and π electrons, take a look at the very first curly arrows ever drawn
The triple bond was added in 1921 by Pease (DOI:10.1021/ja01438a003 ) sharing six electrons.
Again, the three equal lines do not imply identical origins.
The sextuple bond is the largest ever suggested for a real molecule (Mo2 and W2, DOI:10.1039/c2cp43559d ); a "holy grail" is for a stable example in the solid state to be discovered.
For completeness, the maximum bond order for the imaginable periodic table is probably around eight. This is very unlikely to ever be achieved using elements below atomic number ~120.
These basic bond types are assumed to have a layered structure defined by the energy of each type of bond, comprising the first bond (almost always) being a σ type (in fact two are possible), the next two π, the next two δ and the final two φ (s,p,d,f).
The Homo-bond is where a lower layer is absent, as for example a π bond without an underlying σ bond. Thus a homoaromatic bond is a 1-electron rather than 3-electron bond (see next section).
The ionic bond is where the electrons are not shared between two atoms, instead residing on the more electronegative partner.
Very recently a third fundamental type of bond has been introduced (the charge-shift type), which however plays no role in most chemistry degree courses. If you are wondering, an example is that for F2.
The dative bond is deprecated (in organic chemistry). Instead, it is represented as a normal covalent bond, modified by charges on the (two) atoms involved.
Apart from formal covalent bonds, there are also a category of weak bonds (which are not associated with any electron count). These include the hydrogen bond and the dispersion bond. Such bonds do not participate in reactions (in the sense of arrow pushing, see below), but are certainly involved in determining molecular structure, as in e.g. proteins, DNA, and molecular selectivity.
There are other types of bond not involving simply two centres (nuclei) of more relevance to inorganic chemistry.
Other semantic layers have since been added to the representation of these basic types of bonds. One, resonance, is dealt with below. Another, stereochemistry, is also explained later in these notes.
Electronic Resonance
Lewis convention for benzene
Shortly after integer bond orders were introduced, it was recognised that bonds could also have intermediate bond orders, most famously benzene with an order somewhere in-between single and double, due to an electronic phenomenon known as resonance (between two equivalent structures in this case). As well as 2, 4, 6 (8, 10) shared-electron bonds, we now have 1, 3 and 5 shared electron, two-centre (i.e. involving two nuclei) bonds. There are other types as well which we do not mention here. How does our convention represent these?
Armstrong convention for benzeneThe verbose convention uses a resonance (indicated by a double headed arrow) between two equivalent structures. This has the advantage of retaining Lewis bonds, and which in turn allows arrow pushing to be performed with these bonds. Thus benzene is represented with two (Kekule) forms, and it implies six π-electrons in a ring. Sometimes the second valence bond form is omitted, and its presence is therefore implicit (as are the hydrogen atoms).
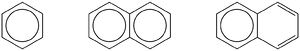
Dotted bond benzeneSometimes this verbose convention is shortened to a structure with an inner circle (a convention introduced by Henry Armstrong, at the precursor to Imperial College, in around 1890 and which he called the centric convention, DOI:10.1039/PL8900600095 ). This convention has two disadvantages. Firstly, it is difficult to use the arrow pushing convention with it. Secondly, it leads to errors. Take for example the representation of naphthalene (middle structure) where each ring is drawn with an inner circle, as first used by Robinson in 1926. This implies two sets of six π-electrons, which is wrong. In fact the correct representation (right) of one circle, and two double bonds, was actually used by Armstrong himself to imply 10 π-electrons (and mis-applied by Robinson). Formally, one would need to also draw a second verbose representation with the circle on the rhs ring. Although circles have their uses (in poly-benzenoid hydrocarbons, as used by Clar), their general use should probably be deprecated.
A third way of representing resonance is the dotted-bond method. This implies that the dotted bond is a 1-electron rather than 2-electron bond (thus used for benzene, it implies a 3-electron bond). It too is imperfect, since it implies some knowledge of the actual electronic distribution in the molecule (something which can only be addressed using molecular orbital theory). The dotted line is also used (on its own, with no other line) to indicate weak bonds, such as hydrogen bonds. Here, there is no implication that such a bond is a 1-electron bond.
Dotted bond acyclic moleculesThe dotted-bond formalism is more often used for other conjugated molecules, such as allyl cation, where the two valence bond forms can be reduced to one with dotted bonds. The required charge is normally placed somewhere in the middle. This however does not mean it is distributed equally amongst all the atoms connected by dotted-bonds. In this case for example, molecular orbital theory would tell that there is no charge on the central atom. Other examples of this formalism would include the nitro group, or a carboxylic acid anion.
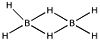
DiboraneThere is one famous exception to the use of Lewis structures. Diborane, which has two bridging hydrogens, is often shown with solid lines for the four bridging bonds. In fact dotted-bonds would also be incorrect, since this is an example of a two-electron-three-centre bond, for which no convenient representation has ever become generally accepted.
With metals in particular, the resonance convention starts to struggle. See here for one challenging example.
The molecule
Conventions in molecule drawing
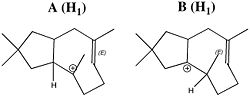
Here we see representations of molecules A-D with a number of implicit conventions, each with different intent.
A, B and C' are drawn as if flat (although of course they are not).
In addition, A and B are drawn with what appears to be 90° angles at two carbon atoms. There is no intent to imply these are real (its probably the result of using templates in the Chemdraw program).
However, there IS deliberate intent to represent a realistic geometry at the alkene (distinguishing between two possible configurational isomers E or Z).
Charges are almost always drawn as being located on one specific atom. Sometimes, a delocalised bond is drawn dashed, and a charge is also associated with this region. That does not however mean that the charge is equally distributed along the length of the dashed bonds (molecular orbital theory would tell that it may be distributed across only a sub-set of the atoms spanned by the dashed bond).
A-C have no hydrogens represented. These atoms are implicit. In this case, the intent is to create a perceptional challenge to the person viewing the diagram (its part of an exam question). The intent might also include avoiding too cluttered a diagram (explicit hydrogens are almost always drawn for a reason).
To identify how many hydrogen atoms are present, you have to apply the rules of (carbon valency) to spot that the two carbon atoms of the alkene together bear only one hydrogen in total, and that the carbon bearing a positive charge has no hydrogens (it is tertiary). A total hydrogen count so obtained (25 if you want to check) is crucial if you want to apply a formula to establish the number of double bond equivalents.
But if you merely want to explain how A converts to B to C, you need to recognise that only one implicit hydrogen needs to be converted to being explicit, as per A (H1) and B (H1). You infer this because the charge involving atoms with implicit hydrogen has moved. In B/C, there is no need to to expose any hydrogens since none are involved in the reaction.
C is drawn in an alternative representation, C1'. Each has a distinct purpose. In an exam for example, C' would be drawn, but perhaps not C, which might be considered a little too helpful.
C is drawn deliberately to match B as closely as possible. No atoms are moved, and only one bond is moved (from the alkene). This has the consequence that two bonds now cross, one over (or under) the other. Our knowledge of perspective forces us to make a decision, and that decision is represented by what is called hidden line removal to indicate which bond obscures the other by passing over it. By specifying which bond is hidden, we immediately impart 3D meanings to three atoms in the structure of C. Notice the diagram now bears stereochemical labels. These were added by the (Chemdraw) program used for the drawing, which recognised stereochemistry at three atoms, and gave each a label (R) or (S).
Now that C has been imparted with 3D information, we can try viewing it from another perspective, C'. This cleans up the structure to reveal the presence of two five-membered rings more clearly. The hidden line is no longer required, but in the process of removing it, we loose the perspective, and ChemDraw no longer has the information it needs to add stereochemical labels. All the hydrogens have again reverted to being implicit.
Conventions in molecule drawingIn D, two new hydrogens (shown in red) are converted from implicit to explicit (neither is the same as the original explicit H). These are recognised (by the human) as being on stereogenic carbon atoms. A decision on their perspective needs to be made, and once done, Chemdraw can again add stereochemical labels, provided stereochemical information has been added to each stereogenic centre (in the form of hashed bonds for the two added hydrogens).
Notice that the bond connecting the two carbons each labelled (S) is a wedge. No explicit hydrogen need be added to generate an unambiguous stereochemical label; it can remain implicit.
But simply adding some perspective to the diagram does not mean the resulting structure is viable. Thus D may or may not be able to sustain normal bond angles; only building a model will tell.
The arrow
The diagram to the right shows the Chemdraw arrow template, with eight rows of arrows in seven columns. The attributes of some of each type of arrow can also be changed by pressing key modifiers (such as option) when invoking a template item. The template mixes meaning and style. The latter is represented by e.g.the size/length/width of an arrow, or its degree of curvature (all of which can be modified by other options). We concentrate here on the meaning (as agreed by convention).
The first row indicates reaction arrows. The first three mean a single step reaction. The last four are retrosynthetic arrows, in which bonds are identified for disconnection in a manner which simplifies the structure (and for which reactions are known for making that bond).
The first three entries in the second row are more reaction arrows. The size of the arrowhead has no meaning.
The last four are curly-arrows, and signify the (formal) movement of an electron pair during a reaction.
The first three entries in row three are a dashed reaction arrow, and serve to indicate a predicted (anticipated) reaction rather than one actually observed (as in this synthesis is not yet complete).
The last four are again curly-arrows, rotating in the opposite sense to the previous ones. The sense of rotation does not normally matter, and the arrow style is selected for layout stylistics.
The first three arrows in rows four and five serve no discernible purpose, other than perhaps for use in molecular orbital diagrams/energy levels, where one arrow would indicate one electron in such an orbital, and two a pair.
The last four arrows in rows four and five are fish-hook arrows, and are used to represent the movement of a single electron in a radical reaction.
The first three arrows in row six are resonance-arrows. They serve to indicate a electronic resonance between two valence-bond structure representations. Unlike reaction arrows, which imply movement of nuclei, these arrows specifically mean no nuclear movement occurs between the two structures.
The last four arrows in row six are double-headed curly arrows. They represent a "lazy" conflation of two separate arrow-pushing steps, the first of which say pushes electrons to e.g. the oxygen atom of a carbonyl group during addition to that group, and the reverse arrow the subsequent elimination from that group. Use of this (heavily semantically loaded) arrow probably means to imply the existence of an intermediate which is not shown in the diagram.
The first three arrows of row seven are negated reaction arrows. They imply a reaction cannot proceed.
The next three arrows of row seven are equilibrium arrows. They connect two species that are in implied equilibrium, and furthermore that the concentration of each is about the same. This arrow can be modified (using the option key) to alter the relative lengths of the two arrows in the pair, to indicate an equilibrium in which the concentrations of each species are unequal.
The last arrow of row seven means rotation about a bond. It has nothing to do with either a reaction or electron arrows.
The first three arrows of row eight are negated reaction arrows. They imply a reaction does not proceed.
The last four arrows have the same meaning as row seven.
Missing from the template above is the Nemesis (inverted roller coaster) arrow, which is often used to indicate a number of consecutive reactions, the intermediates for which are not shown, OR specifically a rearrangement reaction.
The reaction
A typical reaction schematic The reactant is shown with stereochemistry. This will be dealt fully in another item. Here it is taken to mean a single enantiomer, with the (R) configuration at the stereogenic carbon. You are probably expected to predict what the stereochemistry of the product is likely to be.
A reaction arrow has a reagent drawn above it. It might mean in this case that you have the reactant in your flask, and you are adding 1 mol equivalent of reagent to it, from which you might infer that the reactant is always present in excess over the reagent (until the very last drop goes in!).
The temperature of the reactant, dissolved in Et2O as solvent is taken to be -78°C (K is rarely used in this context). This temperature is almost certainly achieved using solid carbon dioxide dissolved in acetone.
A specified temperature also implies a reaction under thermal conditions. In some contexts, this can also be expressed Δ. The heat is probably provided by a mantle if it is above room temperature. The latter is normally designated RT.
Alternative ways of heating are µw (microwave), or ))) (sonication).
Sometimes, light is also used as a condition, and then it is expressed as hν. Rarely is the wavelength of the light quoted directly.
Notice that the methyl group in the reactant is expressed as Me, but as CH3 in the reagent. There is probably no intended semantic meaning to this difference. Equally, the solvent in the top line is expressed as Et2O but it could equally be shown as diethyl ether or even a formula.
Sometimes, a generic (Markush) representation is used, as R (as far as can be told, R was coined around 1880, and stood for Radicle). There should be somewhere a definition of what R might be, as either a single value, say R=Me, or as a range of values, R=Me,Et,Pr. R is always given a shorthand expression, and less commonly as a formula. If multiple Rs are to be used, they are normally expressed as R, R', R".
Notice no time is specified for the reaction. This probably means add the reagent slowly enough to prevent the temperature from exceeding -78°C, but probably also means you might have to consult more detailed instructions to find out how long the reaction should take.
Below the reaction arrow you might have:
Final workup conditions. In this case, it means pour the contents of the flask into (iced) water, and very possibly extract this with diethyl ether.
But it might mean add a second reagent, after you finish adding the first, in a so-called 1-pot procedure. The list may contain more than two items.
The product is labelled A and then in a second step, B. This means
that you have to identify what A and B are. Sometimes, a number is used instead of a letter.
A is enclosed in square brackets, with a hash at the top right. This convention means a transition state, and implies that you are expected to provide a mechanism for the reaction, very probably with arrow pushing. If the hash were to be missing, this implies that A is a transient intermediate, with a lifetime too short to be directly detected by any simple procedure.
The electronic reaction mechanism
Arrow pushing has at least two conventions in common use in text books and elsewhere. A summary of these and others can be found here and you can read about the first ever representation using curly arrows. Mechanistic types themselves can be classified. One of the most general is the classification into patterns of of bond reorganisation, which results in linear reactions, pericyclic reactions and coarctate reactions.
How the conventions arose
Hunter's textbook, 1934These were established largely by textbook authors. The first with a noticeably modern look was Hunter (1934). Here he is explaining using his notation why an ester group is meta-directing towards aromatic electrophilic substitution. The convention of the time was to represent benzene as a simple hexagon, without the additional bonds
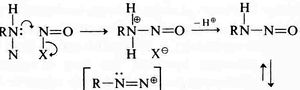
Gould's textbook, 1959 (USA) Gould in 1959 (“Mechanism and structure in organic chemistry”, reviewed DOI:0.1021/ed037p379.2 ) adopted a clearly modern form. In this example, we have a curly arrow starting at the mid-point of a C-H (hydride) bond, and ending at the nucleus of (an electrophilic) carbon atom. His arrows also start at lone pairs rather than negative charges, but at some stage the convention has evolved to dispense with the : indicating a lone pair, and to start the arrow at the charge instead.
Sykes, 1961 (UK) Sykes ("A guidebook to mechanism in organic chemistry" in 1961, reviewed here DOI:10.1021/ed040pA224.1 ) is very similar to Gould, but in his example he shows a nitrogen lone pair heading towards the mid-point of a N-N forming bond, rather than ending at the nucleus of the (electrophilic) nitrogen atom as Gould would have done. Nowadays, we clarify Sykes convention a bit further by adding a dotted line to the forming bond so that the arrow can both start and end on either a lone pair or a line. This dotted line is distinct from dotted or dashed lines used to represent resonance.
Arrow pushing convention 1
This can be referred to as the Sykes convention, and its main points are summarised here:
There are two main types of mechanistic arrows, linear and cyclic (there is a very rare third type, DOI:10.1039/c2cc34026g ). The former have a one clear start and end, the latter can circulate in two directions (clockwise or anticlockwise).
Some reactions may involve using a combination of linear and cyclic arrows (for example the bromination of an alkene or alkene epoxidation by peracid).
The most common mechanism (non-radical) involves just a single arrow either originating or ending at a bond/atom. Normally, no pair of atoms undergo a bond order change between them of more than one.
There are rare exceptions involving two, or even three arrows starting or ending at a bond). The bond order for these can involve changes of 2 or even 3.
Arrows will start at a centre with readily released electrons (nucleophilic for linear reactions). Types of readily released (nucleophilic or nucleus seeking) electron pairs are:
Lone pairs ( : ) associated with an atom. Here, the order of nucleophilicity is C > N > O > F for the first row. The arrow by convention starts at the :.
Bonds. We have to take into account the type of bond.
σ-bonds. Such electron pairs are relatively non-nucleophilic (the s-character of the bond orbital is high) and so only bonds to less electronegative elements can release electrons. Thus a B-H bond can release an electron pair more readily than a C-H bond (in both cases this is called a hydride transfer). Another type of σ-bond which can more easily release electrons is that of cyclopropane (largely because the degree of s-character is lower than a normal σ-bond).
π-bonds. Because these involve only p-AOs (no s-character) they can release electrons relatively easily. Again, this release is easier with less electronegative elements; (B=B) > C=C > C=N > C=O.
δ-bonds, as found in high-bond order metal-metal bonds. Very rarely used in arrow pushing.
Arrows will end at electron accepting sites (electrophiles), to either form a lone pair or a new bond.
The arrow can end at an : associated with an atom. The order of electrophilicity is Halogen > O > N > C > B.
The arrow can end at a bond. Again, a new σ-bond (with high s-character) is a better acceptor of electrons than a π-bond (no s-character), and new bonds associated with more electronegative atoms are the better acceptors. A (formal) positive charge on an atom helps make it a good acceptor (such as a carbocation).
There are four potential combinations of the above rules:
Bond → bond
Bond → lone pair
Lone pair → bond
Lone pair → lone pair. This latter is very rare.
The symmetry of the electrons involved must conform to group theory/symmetry. For example, if the reactant and product of a reaction maintain a plane of symmetry which allows one to distinguish between π- and σ-electrons, one cannot convert a π-pair into a σ-pair during the reaction (or vice versa) if its group-theoretical symmetry has to change. An example of falling foul of this rule is in fact the very first arrows ever pushed in the literature! An elaboration of this rule is used to define whether any particular pericyclic reaction (a reaction with cyclic arrows) is allowed or forbidden.
The convention above makes no attempt to imply symmetry, and as such therefore can result in incorrect mechanisms, as noted above. There are no plans at the moment to add symmetry notation to arrow pushing.
The coordinates of the arrows. This has in the past been very imprecisely defined, but having a precisely defined start and end for each (double-headed, electron pair) arrow could be regarded as being helpful. It is also ascertainable:
Arrows starting or ending at bonds. These coordinates can be computed from the topology of the electron density of either the reactant (the starting point) or the product (the arrow endpoint). Electron density is an experimental observable (using e.g. crystallography) as well as a computable property using quantum mechanics. Its topology (curvatures if you like) can be obtained by appropriate analysis. The key topological property is the bond-critical-point or BCP, which generally can be located at approximately the mid point of the line connecting the two nuclei (its precise position depends on the relative electronegativities).
Arrows starting or ending at lone pairs (:). Here too topological analysis of the electron density can result in defining the centroid of a lone pair, with again precise coordinates.
Practically, no-one is ever going to perform topological analysis of the electron density in order to push arrows! So a good approximation is to assume that a BCP is located at the mid point of a bond and a lone pair is located at an atom (mindful this is NOT coincident with the nucleus, since we know a lone pair has p-character). This approximation leads directly to the Sykes convention.
These points can be summarised in the diagram to the right, involving reaction between butene (as the electron releasing molecule) and HBr (as the electron accepting molecule).
The green dots represent mid-points of bonds (either breaking or making), and more formally correspond to the BCPs described above.
The green : represents a lone pair being formed, more formally corresponding to the lone pair centroid.
A dotted line is drawn to the forming bond. This is not strictly part of the Sykes convention; it can be optionally omitted and left as implied (in much the same way that most hydrogen atoms in molecules are implied).
There are two (optional) red dots also shown. These are another convention which here is explicit, but is often left implicit. One can regard the red dots as the location of hinges, and regard the arrows as rotating about these hinges. A metaphor might be a hinged door, which is opening (bond breaking) by rotating around one hinge, and closing (bond or lone pair forming) by rotating about the next hinge. In this metaphor a covalent bond is a closed door and a lone pair is an open door. Adding these hinges allows one to define a simple checking-rule.
For reactions where no atom undergoes a valency change, the hinges MUST be located on alternating atoms. No two adjacent atoms can have hinges.
An exception might be where linear and cyclic arrows are mixed.
For reactions where one atom undergoes a valency change (the most common examples are 4-valent carbon changing to 2-valent carbon, ie a carbene, or 3-valent nitrogen forming 1-valent nitrene, but it also includes changes in oxidation states of transition metals etc), there must be one occurrence of adjacent atoms (ie bonded atoms) each having a hinge.
Most reactions involve more than one arrow (electron pair). The question can then arise as to the relative timing of the various arrows.
If no explicit intermediate is involved, the arrows are said to be concerted, they all operate at the same time.
Any concerted reaction however need not be synchronous, ie the arrows need not all occur at exactly the same time. Sometimes, the arrows can occur in phases.
To determine either the concertedness or synchronicity of any arrow pushing mechanism is however way beyond our current ability to measure (although there are prospects of doing so). Such properties can be computed, but again doing so requires a very sophisticated calculation. Even if these properties can be ascertained, representing them in the convention shown above is also going to be a challenge. So these attributes are currently not attempted using the conventions above.
Radical reactions. These differ from the electron pair reactions since one arrow is assigned to each electron.
Normally, all the arrows used are single-electron fish-books, but there are some rare cases where both fish-book and normal arrows can be combined (the Birch reduction for example).
In general two fish-hook arrows from different sources will both head off to a bond-mid-point (the BCP of the forming bond).
Although the fish-hook implies an electron spin, there is no convention to ensure that the spin-pairing in any formed new bond is correct (strictly, two fish-hooks of opposite spin should combine).
Because two fish-hook arrows derive from an electron pair, there is no sense of direction (the two arrows head off in opposite directions). Radical arrows tend not to be nucleophilic/electrophilic.
Arrows for reactions involving excited states (photochemistry). These are by and large regarded as beyond the scope of arrow pushing, although one could regard them as triplet state reactions involving fish-hook arrows.
Arrow pushing convention 2
The convention differs from the above in the destination of any bond-forming arrow. This destination is to an atom rather than to a bond.
Worked Example 1: Propene + HBr
The addition of a nucleophile (propene) to an electrophile (HBr) is a typical mechanism involving the acyclic rearrangement of bonds in a reaction to form the most stable carbocationic product. The arrow pushing can serve two (rather different) purposes.
As an accounting mechanism to show the bonds being broken and formed, in such a manner that the normal valencies of the atoms involved are not violated (for example, that the octet rule for carbon is not broken in either reactant or product). In this scheme, the arrows will start either at the mid point of any (nucleophilic) bond or at the centroid of any nucleophilic electron lone pair, and terminate at (approximately) the mid point of any new bond (the electrophilic acceptor) or at a new lone pair (the electrophilic, i.e. electronegative acceptor atom).
An alternative layer is to think in terms of an orbital picture of the reaction, in which a suitable orbital representing the source of electrons (the nucleophile or donor) overlaps with a suitable orbital representing the destination of the electrons (the electrophile or acceptor, which is often an anti-bonding orbital, or a σ* orbital). The former orbital is doubly occupied with two electrons, whilst the latter is empty of electrons, and simple (perturbation) theory indicates that the result of these two orbitals overlapping is an overall stabilisation of the system by a lowering in energy of the occupied orbital (the so-called E(2) perturbation stabilisation). In the model below, the orange phase of the donor is equivalent to the red phase of the acceptor (they are shown like this to help distinguish between them, but you can think that orange≡red and blue≡purple) and the overlap between them results in an overall stabilisation of E(2) = 10.5 kcal/mol (to put this value into context, any value over about 5 kcal/mol is normally thought to have a chemical meaning. For example, anomeric interactions, which you will encounter in more advanced courses, have values of about 15-18 kcal/mol). In this scheme, this orbital picture is in effect simplified by reducing the entire representation to just two arrows. The first arrow starts at the donor alkene π-bond and ends (in this scheme) in the region where the donor overlaps maximally with the acceptor to form a new bond. The second arrow starts at the mid point of the H-Br bond and ends as a lone pair on a newly formed bromide anion.
Overlap of occupied and unoccupied (localised) Orbitals
If you click on the diagram on the right of the orbitals, you will see an animation of the reaction pathway for this process. This is calculated using quantum mechanics and represents the most likely route that the molecules will adopt in reacting. The reaction path shown on the right is the actual calculated energy of the system as it evolves. Notice how the HBr is positioned at the start of the reaction. It is actually forming a hydrogen bonded π-complex to the alkene, but displaced towards the less substituted end of the alkene in what might be called Markovnikov fashion, and leading eventually to the formation of the more stable carbocationic transition state. In fact, the actual reaction path does not stop at the stage shown in the arrow pushing above, but it continues smoothly to form an additional C-Br bond. This last step can be quite dependent on the nature of the solvent the reaction is done in and other factors. In other words, whether an actual carbocation is formed as a species with a significant lifetime, or whether it is immediately quenched by the bromide anion can depend on the reaction conditions.
Both the anti-Markovnikov (left) and Markovnikov (right) reaction pathways are shown here. The latter is 3.5 kcal/mol lower in free energy than the former.
You can also download these and other animations using an iPad by installing the iTunesU app on the device and then activating this hyperlink.
Worked Example 3: Ethene + bromine
This mechanism is deceptively simple, and you will find that several styles have been adopted in text books at various stages. The one shown on the right adopts the convention of ending an electron arrow at a bond rather than at an atom, and three such arrows are used in the first stage of the mechanism (TS1) to produce a bromonium cation and an ion-pair. In this example, an extra molecular of bromine has been employed to stabilise the bromide anion formed. This ion-pair then rearranges its geometry by migrating to the other side of the bromonium cation, and then attacks at the backside (TS2) to produce an anti-periplanar dibromide.
Worked Example 4: Hydroxylamine plus propanone (acetone)
A full explanation of this mechanism is revealed here. The mechanism consists of two stages, the nucleophilic addition to a carbonyl compound to produce a tetrahedral intermediate, and then the breakdown of that intermediate to form an oxide. Only the first stage of this mechanism is shown here.
The spectrum
Conventions in spectroscopy mostly apply to how the data is presented in the experimental section of a report. The most common spectroscopic methods used in organic chemistry are listed below.
The UV/vis spectrum
This is based on the analysis of electronic transition into excited electronic states.
Optical rotation
Comes in two forms, ORP (optical rotatory power) which is the specific rotation of polarised light at a given wavelength (normally the sodium D-line, or 589nm), ORP (Optical rotatory dispersion) is the variation in specific rotation as a function of the wavelength of the polarised light. Rarely reported.
The sign of the measured rotation for any given compound does not necessarily tell you which of the two enantiomers. Even a small change in a remote substituent for the same enantiomer can invert the sign of the measured rotation.
The infra-red (and Raman) spectrum
Infra-red spectroscopy as we know it today started in 1903-5 when Coblentz noticed that characteristic peaks were associated with particular functional groups (DOI:10.1103/PhysRevSeriesI.20.273 ). It is based on analysis of molecular vibrations.
The NMR Spectrum
Crystallography
Stereochemistry
An entire course is devoted to stereochemistry.
The orbital
Orbitals are wavefunctions, (eigenvector) solutions of the Schrödinger equation for either an atom or a molecule. They can be related to the electron density probability distribution in a molecule, and hence give information about the bonding.
Atomic orbitals
These are solutions for an atom, specifically a hydrogen atom. This has only one electron and so an analytic solution is available, which we call atomic orbitals. These can also be transformed into an alternative solution which reflects (anticipates) the eventual geometry at that atom when it becomes a molecule, and these transformed orbitals are called hybrid-atomic-orbitals. Hybrid AOs are normally only constructed from the s and p atomic orbital solutions for the H atom, but they can also be extended to include d AO solutions.
Orbitals for molecules
An orbital describing a molecule has to be obtained as an approximate solution of the Schrödinger equation, and this approximation constructs the wavefunction by combining hydrogenic atomic orbitals according to the variation principle, which produces a solution of minimum energy. In fact there is no unique way of doing this; more than one solution is available, each of which would give an identical electron density distribution for the molecule (the only proper observable). An orbital for a molecule will have an associated energy (an eigenvalue), which might be related to an observable (thus Koopmans' theorem) and coefficients (eigenvectors) which describe the orbital in terms of its atomic orbital constituents. There is a whole family of orbitals, represented by the spectrum below, where to the left we have pure atomic orbitals and to the right delocalised molecular orbitals, with various intermediate solutions:
Atomic orbital → NAO → NHO → NBO/IBO → NLMO → MO
See also DOI:10.1063/1.4802585 for a discussion of the difference between localised and delocalised representations and DOI:10.1021/ct400687b for a brand new suggestion, the Intrinsic bond orbital, which like the NBO, recovers the Lewis classical bonding picture.
Two of the more common types of orbital used for describing molecules are:
The MO (Molecular orbital)
Delocalised Orbital
This is the earliest solution (obtained first in the 1930s) and it represents a delocalised or canonical solution encompassing often many if not all of the atoms in the molecule. This type of orbital, often abbreviated to MO, also reflects the global symmetry of the molecule, ie group theory can be applied to it.
The example shown here is the (highest occupied, or HOMO) molecular orbital calculated for 1-chloro-2-fluoroethane, showing a component between the C-C bond (red), but plenty else.
The Natural bond orbital (NBO)
Localised Orbital
This type, often abbreviated NBO, represents an alternative solution which has "maximum-occupancy character" in localized 1-center and 2-center regions of the molecule. Such a function includes the highest possible percentage of the electron density, ideally close to 2.000, providing the most accurate possible “natural Lewis structure” of ψ. An NBO can be located either in a two-centre bond or a one-centre lone pair. These local solutions map nicely to our concept of functional groups in chemistry, and how we tend to think of reactivity in molecules (Unlike delocalized MOs (whose sprawling forms vary bewilderingly even between closely related systems), NBOs are highly conserved and transferable from one molecular environment to another"). Group theory cannot be applied to this type of orbital.
The example shown here is an NBO calculated for 1-chloro-2-fluoroethane having a component between the C-C bond (red), and almost nothing else.
Applications of orbitals for molecules
The decision of whether to use an MO or an NBO for understanding chemistry depends on whether the property is considered to be a global property or a local property.
A global property encompasses regions of a molecule (or the whole molecule). For example it might be the aromaticity of the molecule (or of a conjugated ring as part of a region of a molecule) or of a cyclic reaction occurring which passes through an aromatic transition state (a pericyclic reaction) or of the ionisation energy of an electron from the whole molecule.
A local property might include consideration of just a functional group involving e.g. two atoms, and how that group might interact with another group, in a manner described as a donor/acceptor interaction. Such an analysis is often used in e.g. conformational analysis.
There is no absolute dividing line between what is a global property and what is a local property; it is often a matter of judgement.
Other useful descriptions of the electron density
Orbitals are not the only function that provides insights into the bonding in a molecule. Two others (thare are many) are listed below
Quantum topology or QTAIM
This is a technique that looks at the curvature (first and second derivatives) of the electron density itself, to identify features known as critical points. These come in four types: nuclear attractors, bond critical points, ring critical points, and cage critical points. The bond-critical point in particular can serve the useful role of providing explicit coordinates for "a bond" as part of e.g. a more formal procedure for arrow pushing.
Electron localisation function(s)
Anomeric effects
This is a method for partitioning electrons into a container or basin with a well defined centroid. These come in two common types; a disynaptic basin (equated with a bond) and a monosynaptic basin (equated with an electron lone pair). Disynaptic basin centroids and bond critical points tend to coincide for most molecules with simple bonding. Monosynaptic basin centroids are useful for providing coordinates for lone pairs for use in e.g. inspecting stereoelectronic alignments or in arrow pushing.
An example of the former is shown on the left, in which the alignment between a lone pair basin (small purple spheres with a halo) and the axis of an adjacent C-Cl bond is computed as -169.0°.