
User:Esc14
Study of Transition States
Introduction
In computational chemistry, when optimizing structures using the potential energy surfaces minima can be found. Those minima correspond to the reactants and products. This means that the energy rises in all direction. In order to obtain the products minimum, the reaction will follow a pathway starting from the reactants minimum and will reach the transition state. The transition state is the highest energy point in this pathway and therefore the energy will decrease in one direction. In order to get the maximum, which correspond to the transition state, the stationery points must first be determined. This is done by using the first derivative of the potential energy. Then by using the second derivative, the sign of the force constant (k) can be determined which will lead to an imaginary frequency if k is negative. By studying the molecular orbitals of those reactions, it can be demonstrated that a Diels-Alder reaction can either be normal or inverse. A normal Diels-Alder reaction is characterized by the energy separation between the HOMO of the diene and the LUMO of the dienophile. On the other hand, an inverse electron demand Diels-Alder reaction is characterized by the energy separation between the LUMO of the diene and the HOMO of the dienophile [1].
[1] L.Lu, S.Yang, Z.Wang, G.Cooks, J. of Mass Spectrometry. Vol 30, 581-594, 1995
Nf710 (talk) 23:09, 4 December 2016 (UTC) Nice intro, good understanding of the stationary points. Some understanding of the method used would be good here.
Reaction of Butadiene with Ethylene
A Diels-Alder reaction is defined as a cycloaddition, which is one of the major classes of pericyclic reactions. The characteristics of those pericyclic reactions is that they have a cycilc transition state and are concerted. The reaction of butadiene with ethene is a perfect example of this type of reactions.

As it is possible to see it on the reaction scheme above, the butadiene C-C bonds correspond to the usual sp2-sp2 bond length for the central bond, which is 1.47 A, and to the usual sp-sp bond length for the terminals bond, which is about 1.37 A. As the cycloaddition proceed, the C-C bonds on the butadiene and the ethene elongate. Right before the transition state the two forming C-C bonds have a length of 2.11 A, which is bigger value than the Van der Waals radius of a Carbon atom (1.7 A). Once the reaction reaches the transition step, the C-C bond shorten until getting to their final length. Those length correspond to sp3-sp3 length (1.54 A), to the sp3-sp2 length (1.50 A) and to the sp-sp length (1.37 A).
Molecular Orbitals of the reactants: Butadiene and Ethene
| Molecular Orbitals of Butadiene | Molecular diagram for the reaction of Butadiene with Ethene | Molecular Orbitals of Ethene | ||||||
|---|---|---|---|---|---|---|---|---|
 |
 |
 |
| |||||
 | ||||||||
 |
 |
|||||||
 | ||||||||
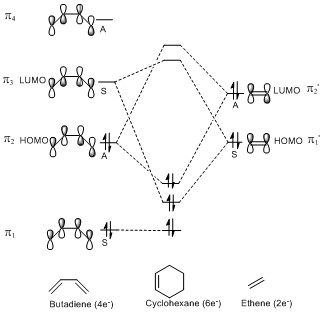
In a thermal reaction, the total number of (4q+2)s and (4r)a components must be odd for the reaction to be allowed. This can be determined by looking at the molecular diagram for the formation of Butadiene/Ethene. The HOMO of butadiene and the LUMO of ethene or the LUMO of Butadiene and the HOMO of Ethene have matching symmetries, respectively asymmetric and symmetric, therefore the reaction is thermally allowed.
Molecular Orbitals of the Transition State
| Bonding Interaction between LUMO of butadiene and HOMO of Ethene | Bonding Interaction between HOMO of butadiene and LUMO of Ethene | Antibonding Interaction between LUMO of butadiene and HOMO of Ethene | Antibonding interaction between HOMO of butadiene and LUMO of Ethene | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
 |
 |
 |
 | ||||||||
As it is possible to observe it on the molecular orbital interactions above, when the LUMO of butadiene (symmetric) interacts with the HOMO of ethene (symmetric) the orbital overlap can either be zero or non-zero according to the bonding interaction. Therefore if the orbitals are bonding, the orbital overlap will be non-zero, whereas if the orbitals are antibonding, the orbital overlap will be zero. This can also be apply to the molecular orbital interactions between the HOMO of butadiene and the LUMO of ethene. The bonding interaction will give a non-zero orbital overlap and the antibonding will give a non orbital overlap. As the symmetries need to be matching for the reaction to be allow, the symmetric-asymmetric interactions don't take place in this reaction. Because the diene provides more in the electron density in the LUMO and the dienophile possesses more electron density in the HOMO, this reaction is considered to be an inverse electron demand Diels-Alder reaction.
23:39, 4 December 2016 (UTC) Terminology not good here, come back
Reaction pathway at the transition state
 |

|
|---|
From the vibration that corresponds to the reaction path at the transition state, it is possible to determine whether this reaction is synchronous or asynchronous. In other terms, determine whether the new bonds are formed simultaneously or whether the formation of one new bonds leads to the formation of the other [1]. As it is possible to see it in the animation above, the butadiene-ethene reaction can be defined as synchronous because the new bonds are formed at the same time.
 |
 |
|---|
As it is possible to observe it on the vibration of the imaginary frequency for the transition state, the bonds bend in a motion that suggests the formation of the cyclohexane transition step. On the other side, the vibration of the lowest positive frequency moves in an asymmetric fashion suggesting that it does not take part in the formation of cyclohexane.
[1] G.Lewars, in Introduction to the Theory and Applications of Molecular and Quantum Mechanics, 2nd Edition, ch.9, p.567
23:45, 4 December 2016 (UTC)This section was done well, excellent knowldge of the theory, you should have measured the bond lengths however
Reaction of Benzoquinone with Cyclopentadiene
The reaction with Benzoquinone and Cyclopentadiene is defined as a [4+2] cycloaddition. It can generate two types of products: the endo and the exo. Using the IRC calculations, it was possible to determine which product is more stable (thermodynamic product) and which is less stable (kinetic product).
Reactants
| HOMO of Cyclopentadiene | LUMO of Cylcopentadiene | ||||||
|---|---|---|---|---|---|---|---|
 |
 | ||||||
| HOMO of Benzoquinone | LUMO of Benzoquinone | ||||||
|---|---|---|---|---|---|---|---|
 |
 | ||||||
Products
| Exo Adduct | Orbital interactions for the Exo and Endo adducts | Endo Adduct |
|---|---|---|
 |
 |
 |
 |
 |
(Watch out that your exo TS orbital diagram shows anti-bonding Tam10 (talk) 11:48, 22 November 2016 (UTC))
Exo adduct
 |
 |
 |
 |
| HOMO of Exo adduct at the transition state | LUMO of Exo adduct at the transition state | ||||
|---|---|---|---|---|---|
According the the HOMO and LUMO of the exo adduct at the transition state, it is possible to determine whether the reaction in normal or inverse. The HOMO shows that the electron density is mostly provided by the cyclopentadiene, which is the diene is this reaction. Whereas the LUMO shows that the electron density mostly comes from the benzoquinone, which is the dienophile in this reaction. As it was stated in the introduction, when the HOMO of the diene reacts with the LUMO of the dienophile it leads to a normal demand Diels-Alder reaction. Therefore, just by looking at the molecular orbitals, it can be demonstrated that the exo reaction between benzoquinone and cyclopentadiene is a normal Diels-Alder.

The HOMO of the exo transition state shown above suggests that secondary orbital interactions could happen.
Nf710 (talk) 10:25, 5 December 2016 (UTC)Good understanding of how the reaction is normal. Be careful as there is no significant SOI in the exo, only in the endo.
Endo adduct
|
|
 |
 |
| HOMO of Endo adduct at the transition state | LUMO of Endo adduct at the transition state | ||||
|---|---|---|---|---|---|
Again, by looking at the HOMO and LUMO of the endo adduct at the transition state, it is possible to determine if the reaction is normal or inverse. In the HOMO, the cyclopentadiene possesses more electron density, whereas in the LUMO the benzoquinone has more electron density. Therefore, the endo reaction can also be defined as a normal demand Diels-Alder reaction. Because the IRC profiles for the endo and exo adducts look very similar, it can be suggested that this cycloaddition is concerted.

The HOMO of the endo transition state shown above suggests that secondary orbital interactions could happen.
Comparaison Study between Endo and Exo pathway
 |
| Energies (in kJ/mol) | ||||
|---|---|---|---|---|
| Exo Reactants | Exo Transition State | Exo Product | Exo Reaction Energy | Exo Reaction Barrier |
| 337.68 | 475.76 | 280.85 | (-56.84) | 138.05 |
| Endo Reactants | Endo Transition State | Endo Product | Endo Reaction Energy | Endo Reaction Barrier |
| 337.68 | 464.04 | 283.82 | (-53.86) | 126.35 |
It can be observed that the exo pathway requires more energy to reach the transition state (138.05 kJ/mol) than the endo pathway. However the overall reaction energy is lower than the overall reaction energy for the endo adduct. More energy is release when the exo product forms, therefore it is consider to be the most stable product (thermodynamic product). Thus, the endo product, which releases less energy, is the kinetic product.
Nf710 (talk) 10:40, 5 December 2016 (UTC) Your Activation energies are correct, but your reaction energies are not , this suggests that you perhaps did not optimise you IRC or used the wrong basis set, other than that this section was done well.
Diels-Alder vs Cheletropic
| Cheletropic product | Diels-Alder product |
|---|---|
 |
 |
 |
 |
Vibrations for the Transition states
| Cheletropic product | Diels-Alder product |
|---|---|
 |
 |
 |
 |
Reaction coordinates for the Cheletropic and Diel-Alder reactions
Unfortunately, the optimization for the Diels-Alder reaction was only done on the exo product. For some unknown reason, the optimization for the transition state at PM6 never went to completion.
(When this happens it's best to upload the log file anyway. There are a number of reasons why the calculation might not have converged Tam10 (talk) 11:48, 22 November 2016 (UTC))
| Diels- Alder product | Cheletropic product | Reaction profile |
|---|---|---|
 |
 |
 |
 |
 |
(Your reaction profile axis should be reaction coordinate, not time! Tam10 (talk) 11:48, 22 November 2016 (UTC))
| Energies (in kJ/mol) | ||||
|---|---|---|---|---|
| Endo Reactants | Endo Transition State | Endo Product | Endo Reaction Energy | Endo Reaction Barrier |
| 210.869 | 247.543 | 12.34 | (-198.529) | 36.67400 |
| Cheletropic Reactants | Cheletropic Transition State | Cheletropic Product | Cheletropic Reaction Energy | Cheletropic Reaction Barrier |
| 210.97 | 256.23 | 6.23 | (-204.74) | 45.26 |
By looking at the energies, it is possible to notice that the reaction barrier is higher for the cheletropic reaction and lower for the Diels-Alder. However, the cheletropic has a lower reaction energy than the Diels-Alder. Therefore, it is possible to conclude that the Cheletropic is the thermodynamically most stable product and the Diels-alder is the kinetic product. Even if the calculations were not run on the two different pathways for the Diels-Alder (exo and endo), it can be predicted that the endo would have a lower transition state than the exo but a higher reaction energy than the exo. As the reaction proceeds the 6-membered ring elongates, until stabilizing to a delocalized resonance structure that is aromatic.
Second cis-butadiene fragment in o-xylylene undergoes a Diels-Alder reaction
| Exo second cis-butadiene | ||
|---|---|---|
 |
 | |
The endo and exo reactions on the second cis butadiene are highly unfavourable because there is not formation of an aromatic system.
| Summary scheme of the different pathways o-Xylylene and So2 reactions |
|---|
 |
Conclusion
In this study on pericyclic reactions using theorical methods, it was discussed how to translate a molecule into a Gaussian input and it was shown how to find specific points on a PES using optimized geometries. The importance of frequency calculations was highlighted when finding a specific point on the PES. It was demonstrated that the kinetic and thermodynamic products can be determined just by looking at the IRC. It was also possible to determine whether Diels-Alder reactions are normal or inverse and whether a reaction is favourable.
Log File
Butadiene with Ethylene
File:EXERCICE 1 BUTADIENE+ETHYLENE PM6 IRC.LOG
Benzoquinone with Cyclopentadiene
File:EXO PM6 MIN.LOG File:EXO PM6 TS.LOG File:EXO PM6 IRC.LOG File:EXO B3LYP TS.LOG File:ENDO PM6 MIN.LOG File:ENDO PM6 TS.LOG File:ENDO PM6 IRC.LOG File:ENDO B3LYP TS.LOG
Diels-Alder vs Cheletropic
File:XYLYLENE-SO2 PM6 MIN.LOG File:XYLYLENE-SO2 PM6 TS.LOG File:XYLYLENE-SO2 PM6 IRC.LOG File:CHELETROPIC PM6 MIN.LOG File:CHELETROPIC PM6 TS.LOG File:CHELETROPIC PM6 IRC.LOG File:XYLYLENE-SO2 ENDO PM6 MIN.LOG