
User:Dk1814
Introduction


A potential energy surface (PES) plots the potential energy of a chemical system (such as the A-B-C system) as a function of internuclear separation for atoms comprising the system. It takes into account interatomic interactions such as Van Der Waals forces using mathematical functions such as the Lennard-Jones (12-6) potential.
A minimum on the potential energy surface has a gradient (first derivative) of zero and a positive second derivative.

A series of minima form a minimum energy path (red bold line in PES) or reaction pathway which allow for chemical reactions to occur. The transition state (TS) is the state with maximum energy along a minimum energy pathway. While the gradient at the transition state is also zero, the second derivative with respect to reaction coordinates is negative.

Nf710 (talk) 22:00, 8 March 2017 (UTC) You have only defined a Minimum and TS in 1D. It is actually in the normal mode basis of 3N-6 dimension.
Vibrational analysis to confirm TS structure

The semi-empirical PM6 & Density Functional Theory-based B3LYP methods were used to compute optimized structures of chemical species. A frequency calculation for an optimised structure generate the normal vibration modes (3N-5 for linear molecules, 3N-6 for non-linear molecules where N is the total number of atoms). Molecular vibrations can be modelled using a Morse Potential which takes into account interatomic attractions/repulsions between adjacent nuclei. For small displacements about the equilibrium distance between two nuclei, the harmonic oscillator approximation can be used: where k is a force constant.
where k is a force constant.
A Taylor expansion about the equilibrium internuclear separation (bottom of potential energy well) yields an expression for the force constant.



A transition state has a negative second derivative along the reaction pathway (negative force constant). Since the frequency of vibration is proportional to the square root of the force constant, a vibration corresponding to the transition state has an imaginary frequency (negative frequency in GaussView).
Nf710 (talk) 22:06, 8 March 2017 (UTC) Very good understanding of the approximations used.
Exercise 1: Diels-Alder reaction between butadiene and ethene

The Diels-Alder reaction is a [4+2] cycloaddition between a s-cis diene and a dienophile which proceeds via a transition state with 6π electrons. Using the frontier orbitals or the highest occupied and lowest unoccupied molecular orbitals (HOMO/LUMO) of the diene and dienophile, the reaction occurs via a suprafacial-suprafacial interaction of orbitals with the correct symmetry.
The thermally allowed Diels-Alder reaction proceeds in 2 ways:
- Overlap of the HOMO of diene with the LUMO of dienophile (Normal electron demand)
- Overlap of the LUMO of diene with the HOMO of dienophile (Inverse electron demand)
| Butadiene | Ethene | |||||||
|---|---|---|---|---|---|---|---|---|
| HOMO |
|
| ||||||
| LUMO |
|
|
Orbitals with the same symmetry label have non-zero overlap integral

(Fv611 (talk) 13:09, 6 March 2017 (UTC) Good MO diagram, but you haven't added symmetry labels for the TS MOs.)
Only orbitals with the same symmetry (symmetric/symmetric or asymmetric/asymmetric) have a non-zero orbital overlap to allow for a reaction to occur. Orbitals with different symmetry have zero orbital overlap; reaction is forbidden. Hence, the interactions between HOMO of butadiene with LUMO of ethene as well as LUMO of butadiene with HOMO of ethene generate 4 molecular orbitals for the transition state. The HOMO and LUMO of the transition state are the bonding and anti-bonding molecular orbitals produced via overlap of the LUMO of butadiene and the HOMO of ethene (due to the smaller energy gap between these orbitals leading to a stronger orbital interaction). Thus, the [4+2] cycloaddition can be characterised as an inverse electron demand Diels-Alder reaction.
(Fv611 (talk) 13:09, 6 March 2017 (UTC) Could have added the formula for orbital overlap.)
- Corresponding MOs for reactants
-
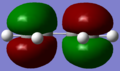 HOMO of butadiene.
HOMO of butadiene. -
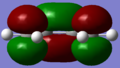 LUMO of butadiene.
LUMO of butadiene. -
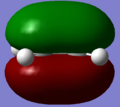 HOMO of ethene.
HOMO of ethene. -
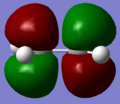 LUMO of ethene.
LUMO of ethene.




- Corresponding MOs for transition state
-
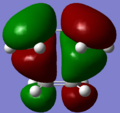 HOMO-1; MO1
HOMO-1; MO1 -
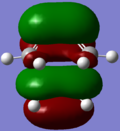 HOMO; MO2
HOMO; MO2 -
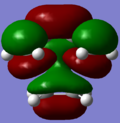 LUMO; MO3
LUMO; MO3 -
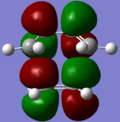 LUMO+1; MO4
LUMO+1; MO4





| C1-C2 | C2-C3 | C1-C6 | C5-C6 | C3-C4 | C4-C5 | |
|---|---|---|---|---|---|---|
| Ethene + Butadiene | 1.47 | 1.34 | 1.34 | - | - | 1.33 |
| Transition State | 1.39 | 1.39 | 1.39 | 2.08 | 2.08 | 1.39 |
| Cyclohexene | 1.34 | 1.50 | 1.50 | 1.54 | 1.54 | 1.54 |
Van Der Waals radius of carbon atom[2]: 1.70 Å
Typical sp3-sp3 C-C bond length: 1.54 Å
Typical sp2-sp2 C=C bond length: 1.34 Å
As the reaction progress, 3 C=C bonds are broken while 2 C-C bonds & 1 new C=C bond forms. The symmetric transition state has 4 carbon-carbon bonds with the same lengths (1.39 Å) corresponding to the partially formed/broken C=C bonds. This bond has a bond order between 1 & 2 and hence an intermediate bond length between a single bond and a double bond.
The distance between the reacting termini carbon atoms in butadiene & ethene (2.08 Å) is significantly smaller than twice the Van Der Waals radius of a carbon atom (3.40 Å), indicating a bonding interaction and the 2 C-C bonds eventually form.


The vibration of the transition state which corresponds to the Diels Alder reaction shows that the formation of the 2 C-C bonds is synchronous and concerted.
Calculations
[Butadiene]
[Ethene]
[TS]
[Cyclohexene]
[IRC]
Exercise 2: Diels-Alder reaction between cyclohexadiene and 1,3-dioxole

The Diels-Alder reaction between cyclohexadiene & 1,3-dioxole results in the formation of exo & endo diastereoisomers via different transition states.
Inverse electron demand Diels-Alder cycloaddition is synchronous
1,3-dioxole is a relatively electron-rich dienophile due to the donation of lone pairs on the oxygen atoms into the π/π* molecular orbitals of the alkene. This raises the energy of both the HOMO and LUMO of the dienophile. The HOMO lies closer in energy to the LUMO of cyclohexadiene and the Diels-Alder reaction is driven by an inverse electron demand.

- MOs for exo TS
-
 HOMO-1
HOMO-1 -
 HOMO
HOMO -
 LUMO
LUMO -
 LUMO+1
LUMO+1




- MOs for endo TS
-
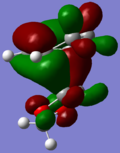 HOMO-1
HOMO-1 -
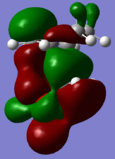 HOMO
HOMO -
 LUMO
LUMO -
 LUMO+1
LUMO+1




 |
 |
 |
 |
The concerted Diels-Alder reaction between cyclohexadiene and 1,3-dioxole involves the synchronous formation of 2 C-C bonds to generate a new 6-membered ring in the product.
Secondary orbital interactions stabilise the endo transition state
By comparing the HOMO of the exo and endo transition states, there is an additional interaction between the O2p orbitals and the LUMO of the diene. Both transition states suffer from similar steric repulsions - Van der Waals strain due to the distance between neighboring C & O atoms (~ 3 Å) being less than the sum of their Van der Waals radii; Van der Waals radius of Oxygen is 1.52 Å[2], sum of radii = 3.22 Å. Hence, the secondary orbital interaction stabilises the endo TS and lowers the activation energy for formation of the endo product.
-
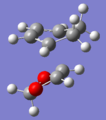 endo TS
endo TS -
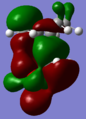 endo TS HOMO
endo TS HOMO -
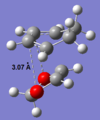 Van der Waals repulsion between C & O
Van der Waals repulsion between C & O


-
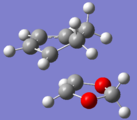 exo TS
exo TS -
exo TS HOMO
-
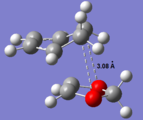 Van der Waals repulsion between C & O
Van der Waals repulsion between C & O



| exo- | endo- | |
|---|---|---|
| Reactants | -1313780.63 | -1313780.63 |
| Transition State | -1313614.33 | -1313622.16 |
| Product | -1313845.75 | -1313849.37 |
The endo product is kinetically favored (endo TS 7.83 kJ mol-1 lower in energy than exo TS) due to stabilising secondary orbital interactions between oxygens and the diene. It is thermodynamically favored due to less destabilising steric clashes; the exo product is relatively congested on its top face and suffers from steric repulsions between the ethylene bridge and the dioxane ring.
-
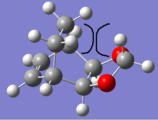 Exo product suffers from steric congestion on its top face
Exo product suffers from steric congestion on its top face -
 Endo product has less steric clashes
Endo product has less steric clashes


Calculations
[Cyclohexadiene] [1,3-dioxole] [Endo TS] [Endo product] [Exo TS] [Exo product] [Endo Diels-Alder IRC] [Exo Diels-Alder IRC]
Nf710 (talk) 22:21, 8 March 2017 (UTC) Very Nice diagrams. But you haven't actually given the energy of reaction or activation energies. just the raw energies. They mean nothing unless they are calculated relatively.
Exercise 3: Reactions between o-xylylene and sulphur dioxide: Hetero Diels-Alder vs Cheletropic

Asynchronous Diels-Alder cycloaddition
 |
 |
 |
 |
 |
 |
The Diels-Alder reaction between ortho-xylylene and SO2 involves the asynchronous formation of C-O and C-S bonds to generate a 6-membered ring. The C-O bond forms first, followed by the C-S bond. The cheletropic reaction involves synchronous formation of 2 C-S bonds to generate a 5-membered ring.

| exo- | endo- | cheletropic | |
|---|---|---|---|
| Reactants | 154.37 | 154.37 | 154.37 |
| Transition State | 241.75 | 237.77 | 260.09 |
| Product | 57.92 | 56.32 | -0.01 |
(You've mixed up exo and endo products Tam10 (talk) 12:45, 8 March 2017 (UTC))
The endo product is kinetically and thermodynamically favored over the exo product. The lower activation energy is likely due to additional interactions between O2p orbitals and the diene HOMO stabilising the endo transition state. While both products are similar in steric requirement, the endo product is likely to be stabilized by through space bonding interactions involving the O2p orbitals.
-
 Endo TS
Endo TS -
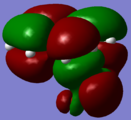 HOMO - Secondary orbital interaction with O2p
HOMO - Secondary orbital interaction with O2p -
 Exo TS
Exo TS -
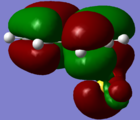 HOMO - No interaction with exo oxygen
HOMO - No interaction with exo oxygen




-
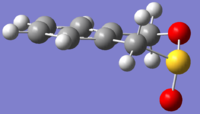 Endo product
Endo product -
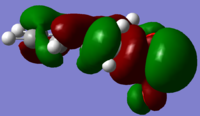 HOMO - Additional orbital overlap with O2p
HOMO - Additional orbital overlap with O2p -
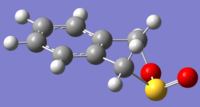 Exo product
Exo product -
 HOMO - No interaction with exo oxygen
HOMO - No interaction with exo oxygen




The cheletropic product is the most thermodynamically stable. Due to the larger S atom (VDW radius of 1.8 Å compared to 1.7 Å for carbon), the 5-membered sulfolane heterocycle is likely to suffer from less ring strain than the 6-membered heterocycles in the Diels-Alder products. However, it forms the slowest (highest activation energy); this is likely due to destabilizing repulsions between the oxygen lone pairs as the oxygen atoms are closer in proximity in the transition state (O-S-O angle decreases from 120 o to ~109 o as the reaction progresses).
Instability of o-xylylene

(You should try some calculations on this to confirm Tam10 (talk) 12:45, 8 March 2017 (UTC))
ortho-xylylene is highly unstable and undergoes electrocyclic ring closure to generate the aromatic product, benzocyclobutene[3].
Both reaction pathways with sulphur dioxide generate an aromatic product with a phenyl group. As the reaction progresses, 6π electrons become delocalised in the 6-membered ring and all 6 C-C bonds are equivalent (bond lengths of 1.4 Å; partial double bond character). The gain in aromaticity drive the reactions thermodynamically.
Alternate Diels-Alder reaction

ortho-xylylene has an endocyclic diene component which can undergo a Diels-Alder reaction with sulphur dioxide.
 |
 |
 |
 |

| exo- | endo- | |
|---|---|---|
| Reactants | 154.37 | 154.37 |
| Transition State | 275.82 | 267.99 |
| Product | 176.71 | 172.26 |
A Diels-Alder reaction at the second cis-butadiene is both kinetically & thermodynamically unfavourable due to the loss of conjugation as the reaction progresses (exocyclic C=Cs no longer planar with partially broken endocyclic C=Cs in the transition state) and lack of aromaticity in the product (localised C=C in 6-membered ring leads to a higher energy product relative to reactants).
Calculations
[o-xylylene] [SO2] [Endo TS] [Endo product] [Exo TS] [Exo product] [Cheletropic TS] [Cheletropic product] [Other exo TS] [Other exo product] [Other endo TS] [Other endo product] [Endo Diels-Alder IRC] [Exo Diels-Alder IRC] [Cheletropic IRC] [Other endo Diels-Alder IRC] [Other exo Diels-Alder IRC]
Conclusion
Two different computational methods were used to investigate the transition state as well as kinetic & thermodynamic products for pericyclic reactions. While the semi-empirical PM6 method utilises experimental data and allows for faster calculations, the Density Functional Theory based B3LYP/6-31G(d) method yields better optimization by using a higher split-valence basis set to generate molecular orbitals.
The concerted Diels-Alder reaction between butadiene and ethene was shown to proceed via a symmetrical transition state with synchronous C-C bond formation to generate cyclohexene.
For the inverse electron demand Diels-Alder reaction between cyclohexadiene and 1,3-dioxole, the endo adduct was the kinetic and thermodynamic product due to electronic (secondary orbital overlap) & steric (less steric clashes) effects.
Of the two feasible pericyclic reactions between o-xylylene and sulphur dioxide, the cheletropic reaction yielded the thermodynamic product (more stable 5-membered sulfolene ring) while the kinetic product was the endo Diels-Alder adduct (possible secondary orbital overlap). The hetero-Diels-Alder reaction involving the endocyclic diene was shown to be kinetically and thermodynamically unfeasible.
As transition states are difficult to analyze in conventional experiments as they are not isolated, computational chemistry is extremely useful for modelling the progression of chemical reactions and providing valuable information about transition states.