
Rep:Module1:louiso14
Module 1
The main aim of this experiment was to become familiar with the programs used for molecular modelling. These included Chembio3D and Gaussian. Molecular modelling uses molecular mechanics to predict the geometries and energy of 3D molecules.
Part 1: Modelling Using Molecular Mechanics
The program used for this exercise was Chembio3D and the method employed was MM2.
The Hydrogenation of Cyclopentadiene Dimer
This part of the exercise involves looking at the dimerisation of cyclopentadiene which occurs via a cycloaddition reaction and which can result in either an endo or exo product. The hydrogenation of the endo product is also investigated.
The dimerisation of cyclopentadiane can either be thermodynamically controlled or kinetically controlled. The thermodynamic product is more stable whereas the kinetic product is formed the fastest even though it is not the most thermodynamically stable. The activation energy required to form the thermodynamic product is greater than that required to form the kinetic product and so if a reaction proceeds via a pathway where the activation energy is lower then the resultant product would be the kinetic one. The kinetic product is able to revert back to the starting molecule and then to the thermodynamic product, but once the thermodynamic product is formed it is not able to revert back.
The 3D representations of the molecules under investigation are shown in the gallery below in their lowest energy forms,as generated by the MM2 minimization.
-
 exo product
exo product -
 endo product
endo product -
 Molecule 3
Molecule 3 -
 Molecule 4
Molecule 4




The data returned by Chembio3D for each molecule is shown in the table below. The data corresponds to the geometry optimized structures of each molecule.
| Energy type | Exo product | Endo product | Molecule 3 | Molecule 4 |
|---|---|---|---|---|
| E/Kcalmol-1 | E/Kcalmol-1 | E/Kcalmol-1 | E/Kcalmol-1 | |
| Stretch | 1.2852 | 1.2520 | 1.2368 | 1.1334 |
| Bend | 20.5883 | 20.8466 | 18.6334 | 13.0199 |
| Stretch-Bend | -0.8376 | -0.8370 | -0.7466 | -0.5647 |
| Torsion | 7.6718 | 9.5139 | 12.8915 | 12.4289 |
| Non-1,4 VDW | -1.4354 | -1.5557 | -1.3635 | -1.3390 |
| 1,4 VDW | 4.2329 | 4.3341 | 6.0549 | 4.4346 |
| Dipole/Dipole | 0.3778 | 0.4490 | 0.1632 | 0.1410 |
| Total energy | 31.8830 | 34.0015 | 36.8696 | 29.2540 |
Dimerisation
The major contribution to the difference in the observed energies of the endo and exo products is the torsion energy, as it can be seen that the torsional strain experienced by the endo product is greater and this leads to its higher overall energy. This torsional strain is mainly caused by the new carbon-carbon formed in the dimer. The majority of the energy within both molecules is from the 'bend' and this is when the the bond angles within a molecule deviate from ideality. This deviation from ideality is due to steric intercations.
From the results obtained it appears that the exo product is lower in energy by 2.12 kcal/mol and therefore more thermodynamically stable, yet it is the endo product which is accepted as the major product. This must mean that the the endo product is formed via the kinetic pathway and that it is the kinetic product and therefore, that the reaction is kinetically, not thermodynamically controlled. The endo transition state is more stable and this is why the reaction proceeds via the lowest energy pathway. The endo transition state is lower in energy due to secondary orbital interactions and these interactions are those which have a favourable overlap in the transition state, yet they are not involved in any bonding. They therefore lower the total energy of the transition state, allowing the reaction to proceed via the lowest energy pathway.
Hydrogenation of the endo product
Molecule 4 is thermodynamically more stable than molecule 3 and by 7.62 Kcal/mol. Since molecule 4 is thermodynamically more stable it can be regarded as the major hydrogenation product. The biggest contribution to the difference in observed energies is due to the bend energy, as for molecule 4 it is 5.61Kcal/mol lower than in molecule 3. This means that the bond angles in molecule 4 are nearer the desired ones. Observing the structures of the two hydrogenated molecules gives evidence as to why molecule 4 is favoured and this is because the strain created by the double bond closest to the bridging methyl is greater than the strain caused by the other double bond. Therefore, in molecule 4 the double bond closest to the bridging methyl group has been hydrogenated and so the relief from the strain is greater resulting in a lower overall energy.
Stereochemistry of nucleophilic additions to a pyridinium ring
The following two reactions are examples of nucleophilic addition reactions and molecular mechanics will be used to confirm why the absolute stereochemistry seen occurs.
Reaction of a Grignard and a Proline derivative
The overall reaction is shown below. It involves the grignard attack on the 4 position of the pyridine ring, which results in the methyl group being added at the 4 position with the stereochemistry shown.This reaction has also been previously studied in the literature[1].

Firstly an attempt was made to model molecule 5 with the grignard reagent but this did not work due to Chembio3D not recognizing the magnesium atom belonging to the grignard reagent. This is because Chembio3D lacks the relevant force field parameters to deal with metal atoms. Molecule 5 was then modelled on its own.
 |
 |
The geometry optimized structure of molecule 5 is shown to the right. The total energy for the structure was 43.115 kcal/mol and this was the lowest energy conformation found, with the carbonyl group above the the pyridine ring by 11 degrees. When the angle was increased the overall energy of the molecule also increased. Taking into account the fact that the carbonyl oxygen points above the ring, this helps to explain why the methyl group is added to the top face in the reaction. This is because the mechanism involves the magnesium atom of the grignard coordinating to the carbonyl oxygen and so the methyl group is added above the plane of the ring and the absolute stereochemistry results.
Reaction of Analine with a Pyridinium ring

The scheme for the reaction of analine and a pyridinium ring is shown above. Molecular mechanics is used to rationalize why the observed stereochemistry occurs.
 |
Molecule 7 was found to have a total energy of 63.1631 kcal/mol and this was for the optimized form. As the diagram to the left shows, the optimized form had the carbonyl oxygen pointing below the ring at an angle of 17 degrees. This helps to explain why the stereochemistry of the reaction is seen and this because analine is a rather large nucleophile and so quite sterically hindered. It will therefore approach the pyridinium ring from above rather than below. Other considerations are that the analine has a lone pair of electrons on the nitrogen which are likely to be repulsed by the lone pairs of the carbonyl oxygen and that the analine will not coordinate to the carbonyl oxygen in the same way that the magnesium grignard did earlier. Considering these points explains why the NHPh group adds to the top face of the ring.
Possible Improvements
The main limitation with using the MM2 method is that it doesn't take into account the electron density within a molecule, it only considers simple interactions such as dipoles and sterics. An improvement would be to use to use the MOPAC/PM6 function or to use Gaussian.
Synthesis of Taxol
This exercise involves looking at a key intermediate in the synthesis of Taxol. The key intermediate can either be synthesized with the carbonyl group pointing up or down and molecular mechanics will be used to identify the most stable isomer. The two isomers are an example of atropisomerism and this due to the restricted rotation of the ring. The structures of the two isomers can be seen below.
 |
 |
The total energy of molecule 9 was recorded as 48.889 Kcal/mol whereas the total energy of molecule 10 was recorded as 44.3282 kcal/mol. From the results of molecular mechanics it appears that molecule 10 with the carbonyl group pointing down is the most stable form. The largest contribution to the difference in the observed total energies was the bending contribution. The bending energy of molecule 9 was greatest and this is when the carbonyl group is facing up and causes the bond angle of the sp2 hybridised carbonyl carbon to deviate from its ideal angle of 120 degrees as the observed bond angle is in fact lower.
It should also be noted that the total energies quoted are of the lowest energy forms and these low energies were acheived by the arranging of the cyclohexane ring component into a chair form in both cases. For example with molecule 9, when the six membered ring was in a boat conformation the total energy was 54.43 kcal/mol, which lowered considerably when the boat form was converted to a chair form (48.889 kcal/mol).
The alkene reacts slowly because it is regarded as a hyperstable olefin[2]. This is where the hydrogenated product is higher in energy than the starting alkene. The starting alkene is more stable because the molecule experiences less strain and this is known as negative olefin strain. The strain experienced by taxol when it reacts is probably even greater due to the presence of the bridging methyl group next to the double bond.
Part2:Modelling using Semi-empirical molecular orbital theory
This section involves using the MOPAC/PM6 method and the Gaussian program. This is so that molecular orbitals can be studied.
Regioselective addition of Dichlorocarbene
Part 1

Molecule 12 was first minimized using the MM2 method and the total energy returned was 17.8990 kcal/mol. The MOPAC/PM6 interface was then used to calculate the molecular orbitals of dichlorocarbene. It was also noted that when the MOPAC interaface was used to minimize the energy of molecule 12 and the molecule became less linear.
The molecular orbitals of molecule 12 calculated by the MOPAC interface are shown below.
-
 molecule 12 lumo+1
molecule 12 lumo+1 -
 molecule 12 lumo
molecule 12 lumo -
 molecule 12 homo
molecule 12 homo -
 molecule 12 homo-1
molecule 12 homo-1




From the HOMO molecular orbital diagram it can be seen that the double bond anti(endo) to the C-Cl bond has much greater electron density than the exo double bond. The endo double bond is therefore more likely to undergo electrophilic attack than the exo double bond[3]. Also from looking at the HOMO-1 and LUMO+1 orbitals it appears that from the correct symmetry seen, the HOMO-1 orbital can donate electron density into the LUMO+1 orbital and this corresponds to the exo C=C bond donating electron density into the C-Cl antibonding orbital. This weakens the C-Cl bond.
The C=C double bond anti to the C-Cl bond in molecule 12 was then replaced with a C-C single bond and the molecular orbitals were calculated. This molecule is referred to as molecule 13. Molecule 13 was found to be higher in energy, around 22.3 kcal/mol.
-
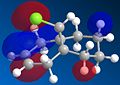 molecule 13 lumo+1
molecule 13 lumo+1 -
 molecule 13 lumo
molecule 13 lumo -
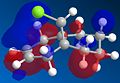 molecule 13 homo
molecule 13 homo -
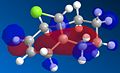 molecule 13 homo-1
molecule 13 homo-1




The largest difference in the molecular orbitals of molecules 12 and 13 can be seen by looking at the HOMO orbitals, as the HOMO of molecule 13 clearly shows a large amount of electron density over the exo double bond and not over the newly formed carbon-carbon single bond, which in molecule 12 was a double bond and contained a large amount of electron density. One would expect the C-Cl bond to be stronger in molecule 13 than in molecule 12 because of no donation of electron density from the HOMO-1 to the LUMO+1.
Part 2
Gaussian input files for both molecules 12 and 13 were created and their vibrational frequencies were analysed.
| Molecule 12 | Molecule 13 | |
|---|---|---|
| Bond | vib. freq./cm-1 | vib. freq./cm-1 |
| C=C (exo) | 1757.2 | 1758.3 |
| C=C (endo) | 1737.7 | n/a |
| C-Cl | 770.2 | 775.8 |
The C-Cl vibrational frequency of molecule 13 is larger than that of molecule 12, which means that the C-Cl bond in molecule 13 is stronger, as previously stated. This difference is due to the pi system of the C=C bond donating electron density into the antibonding orbital of C-Cl bond, which destabilises the C-Cl bond and only occurs in molecule 12. This also why the exo double bond in molecule 12 is stronger than the endo double bond. Molecule 13 only has one double bond and so only one vibrational frequency is displayed.
Structure based miniproject using DFT-based Molecular orbital methods: Spectroscopic study of a fused pyran-γ-lactone
Fused pyran-γ-lactones are common structural motifs in many naturally occurring products of which some possess biological activity. The fused-γ-pyran lactone is synthesized via a Pd-Thiourea-Catalyzed Alkoxycarbonylative Annulation. The synthesis is taken from a journal which successfully synthesizes the fused-γ-pyran lactone [4]. The fused-γ-pyran lactone under investigation has two possible isomers, where two hydrogen atoms can either be trans or cis to each other. In this study the experimentally obtained 13C NMR spectra will be compared to calculated spectra. The reaction producing the two isomers is shown below:

The cis and trans isomers were then drawn in Chembio3D and the MM2 function was performed. Their optimized structures are shown below along with Jmols of when they were optimized.
 |
The total energy of the cis isomer was 16.6202 kcal/mol whereas the trans isomer's total energy was 17.1880 kcal/mol. The total energies reported show that the cis isomer is the most thermodynamically stable and this is reflected in the percentage yields obtained in the journal as the cis isomer had a yield of 83% and was the major product, whereas the trans isomer only had an 11% yield.
Diagrams of numbered carbon atoms for the two isomers, which correspond to the atoms listed in the NMR data tables.
-
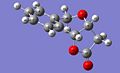 cis isomer
cis isomer -
 trans isomer
trans isomer


A comparison between the experimentally otained and predicted NMR spectra will now be made.
Cis-isomer 13C NMR spectra
The NMR spectra for the cis-isomer can seen below. Please click on the images to enlarge them.
-
![Experimentally obtained 13C NMR of cis-isomer[5]](/images/thumb/3/32/Lo.Real_nmr_cis.JPG/120px-Lo.Real_nmr_cis.JPG) Experimentally obtained 13C NMR of cis-isomer[5]
Experimentally obtained 13C NMR of cis-isomer[5] -
 Predicted 13C NMR of cis-isomer
Predicted 13C NMR of cis-isomer
![Experimentally obtained 13C NMR of cis-isomer[5]](/wiki/File:Lo.Real_nmr_cis.JPG)

| Carbon atom | Lit. value/ppm | Calculated value/ppm | difference/ppm |
|---|---|---|---|
| 13 | 37.6 | 36.6 | 1 |
| 10 | 66.7 | 63.1 | 3.6 |
| 8 | 73.3 | 71.8 | 1.5 |
| 7 | 75.5 | 76.6 | 1.1 |
| 6 | 124.4 | 122.4 | 2.4 |
| 2 | 127.6 | 134.3 | 2.4 |
| 1 | 127.7 | 126.1 | 1.6 |
| 3 | 129.3 | 126.7 | 2.6 |
| 4 | 130.7 | 127 | 3.7 |
| 5 | 135.1 | 136 | 0.9 |
| 12 | 174.7 | 175.9 | 1.2 |
The average difference in ppm is 2.08 ppm. This therefore shows that the calculated NMR data compares well to the literature values. The calculated value for carbon number 12 was corrected, as it is a carbonyl carbon of an ester. The original value calculated was 170.6 ppm so the correction brought the value a lot closer to the literature value.
Trans-isomer 13C NMR spectra
The NMR spectra for the trans-isomer can seen below. Please click on the images to enlarge them.
-
![Experimentally obtained 13C NMR of trans-isomer[6]](/images/thumb/2/2a/Lo.Real_nmr_trans.JPG/120px-Lo.Real_nmr_trans.JPG) Experimentally obtained 13C NMR of trans-isomer[6]
Experimentally obtained 13C NMR of trans-isomer[6] -
 Predicted 13C NMR of trans-isomer
Predicted 13C NMR of trans-isomer
![Experimentally obtained 13C NMR of trans-isomer[6]](/wiki/File:Lo.Real_nmr_trans.JPG)

| Carbon atom | Lit. value/ppm | Calculated value/ppm | difference/ppm |
|---|---|---|---|
| 13 | 36.1 | 36.7 | 0.6 |
| 10 | 69.5 | 68.5 | 1.4 |
| 8 | 75.9 | 75.1 | 0.8 |
| 7 | 78.1 | 76.5 | 1.6 |
| 3 | 123.3 | 120.8 | 2.5 |
| 6 | 124.4 | 121.3 | 3.1 |
| 2 | 127 | 123.1 | 3.9 |
| 1 | 128.1 | 124.3 | 3.8 |
| 4 | 132.8 | 130.8 | 2 |
| 5 | 133.2 | 131.1 | 2.1 |
| 12 | 172.6 | 173.3 | 0.7 |
The average difference in ppm is 2.04 ppm, so the calculated data compares well to the literature. Again the carbonyl carbon of the ester had to be corrected and this also brought the calculated value closer to the literature value.
Therefore, the average difference observed in ppm for both isomers is roughly the same and it is a relatively low value. This suggests that the GIAO method has worked well and has quite accurately predicted the 13C NMR of both the cis and trans isomers.
References
- ↑ A. G. Shultz, L. Flood and J. P. Springer, J. Org. Chemistry, 1986, 51, 838. DOI:10.1021/jo00356a016
- ↑ 'Evaluation and prediction of the stability of bridgehead olefins' - Wilhelm F. Maier, Paul Von Rague Schleyer -Journal of the American Chemical Society 1981 103 (8), 1891-1900
- ↑ B. Halton, R. Boese and H. S. Rzepa, J. Chem. Soc., Perkin Trans. 2, 1992, 447-8. DOI:10.1039/P29920000447
- ↑ Diversity-Oriented Synthesis of Fused Pyran γ-Lactones via an Efficient Pd−Thiourea-Catalyzed Alkoxycarbonylative Annulation,Zhengtao Li, Yingxiang Gao, Zhaodong Jiao, Na Wu, David Zhigang Wang, Zhen Yang, Organic Letters 2008 10 (22),5163-5166.DOI:10.1021/ol802115u
- ↑ Diversity-Oriented Synthesis of Fused Pyran γ-Lactones via an Efficient Pd−Thiourea-Catalyzed Alkoxycarbonylative Annulation,Zhengtao Li, Yingxiang Gao, Zhaodong Jiao, Na Wu, David Zhigang Wang, Zhen Yang, Organic Letters 2008 10 (22),5163-5166.DOI:10.1021/ol802115u
- ↑ Diversity-Oriented Synthesis of Fused Pyran γ-Lactones via an Efficient Pd−Thiourea-Catalyzed Alkoxycarbonylative Annulation,Zhengtao Li, Yingxiang Gao, Zhaodong Jiao, Na Wu, David Zhigang Wang, Zhen Yang, Organic Letters 2008 10 (22),5163-5166.DOI:10.1021/Ol802115U