NH3 (Ammonia) Molecule
| Information
|
|
|
| NH3 (Ammonia) Molecule
|
| Calculation Details
|
| Method
|
Basis Set
|
Point Group
|
| RB3LYP
|
6-31G(d,p)
|
C3v
|
| Calculation Results
|
| E(RB3LYP) /a.u
|
RMS Gradient /a.u.
|
| -56.55776873
|
0.00000485
|
| Click here for the log file.
|
Item Value Threshold Converged?
Maximum Force 0.000004 0.000450 YES
RMS Force 0.000004 0.000300 YES
Maximum Displacement 0.000072 0.001800 YES
RMS Displacement 0.000035 0.001200 YES
Optimized Bond Length : 1.01798 Angstrom
Optimized Bond Angle : 105.741°
| Vibration and Charge Screenshots
|
|
|
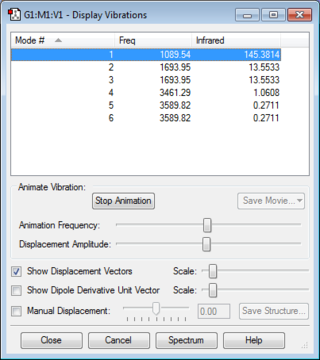
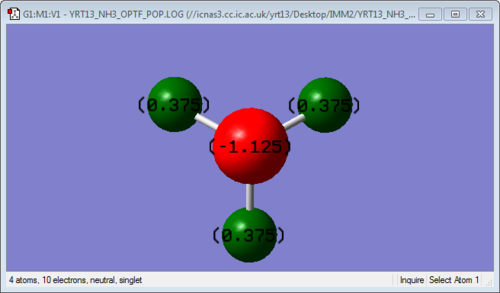
Modes Expected from 3N-6 Rule: 6
Degenerate Modes: Modes 2 and 3 are degenerate. Modes 5 and 6 are degenerate too.
Bending Vibration Modes: 1, 2 and 3
Stretching Vibration Modes: 4, 5 and 6
Highly Symmetric Mode: 4
"Umbrella" Mode: 1
Expected Number of Experimental Spectrum Bands of Gaseous Ammonia: 3
N2 & H2 Molecules
N2 Molecule
| Information
|

|
|
| N2 Molecule
|
| Calculation Details
|
| Method
|
Basis Set
|
Point Group
|
| RB3LYP
|
6-31G(d,p)
|
D∞h
|
| Calculation Results
|
| E(RB3LYP) /a.u
|
RMS Gradient /a.u.
|
| -109.52412868
|
0.00000060
|
| Click here for the log file.
|
(N2) Item Value Threshold Converged?
Maximum Force 0.000001 0.000450 YES
RMS Force 0.000001 0.000300 YES
Maximum Displacement 0.000000 0.001800 YES
RMS Displacement 0.000000 0.001200 YES
| Vibration and Charge Screenshots (for N2)
|

|

|
There are no negative frequencies. Intensity of infrared is zero as there is no change in dipole moment (due to symmetric stretching). Furthermore, since the difference in electronegativity of both atoms is zero (homonuclear diatomic molecule), both atoms have zero charge.
H2 Molecule
| Information
|

|
|
| H2 Molecule
|
| Calculation Details
|
| Method
|
Basis Set
|
Point Group
|
| RB3LYP
|
6-31G(d,p)
|
D∞h
|
| Calculation Results
|
| E(RB3LYP) /a.u
|
RMS Gradient /a.u.
|
| -1.17853936
|
0.00000017
|
| Click here for the log file.
|
(H2) Item Value Threshold Converged?
Maximum Force 0.000000 0.000450 YES
RMS Force 0.000000 0.000300 YES
Maximum Displacement 0.000000 0.001800 YES
RMS Displacement 0.000001 0.001200 YES
| Vibration and Charge Screenshots (for H2)
|
|
|
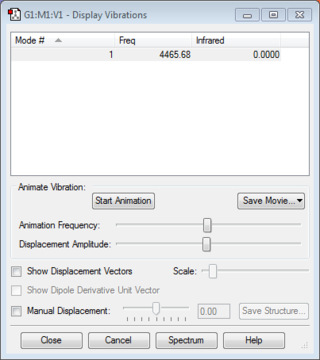
Similarly, there are no negative frequencies. Intensity of infrared is zero as there is no change in dipole moment (due to symmetric stretching). Furthermore, since the difference in electronegativity of both atoms is zero (homonuclear diatomic molecule), both atoms have zero charge.
Reaction Energies
For the reaction N2 + 3H2 -> 2NH3:
E(NH3)= -56.55776873 a.u.
2*E(NH3)= -113.11553746 a.u.
E(N2)= -109.52412868 a.u.
E(H2)= -1.17853936 a.u.
3*E(H2)= -3.53561808 a.u.
ΔE=2*E(NH3)-[E(N2)+3*E(H2)]= -0.05579070 a.u.= -146.48 kJ/mol
Therefore, the energy change associated with the conversion of hydrogen and nitrogen gas into ammonia gas is -146.48 kJ/mol
The ammonia product is more stable as the reaction is endothermic.
The experimental enthalpy change (ΔH = −92.4 kJ/mol) is quite different from the energy change calculated above. This tells us that the use of simple DFT methods to calculate thermodynamic data is insufficient.
CH4 (Methane) Molecule
| Information
|

|
|
| CH4 (Methane) Molecule
|
| Calculation Details
|
| Method
|
Basis Set
|
Point Group
|
| RB3LYP
|
6-31G(d,p)
|
Td
|
| Calculation Results
|
| E(RB3LYP) /a.u
|
RMS Gradient /a.u.
|
| -40.52401404
|
0.00003263
|
| Click here for the log file.
|
(CH4) Item Value Threshold Converged?
Maximum Force 0.000063 0.000450 YES
RMS Force 0.000034 0.000300 YES
Maximum Displacement 0.000179 0.001800 YES
RMS Displacement 0.000095 0.001200 YES
Vibration Properties
| Vibration Properties
|
| Mode
|
Frequency/cm-1
|
Intensity
|
Comments
|
| 1
|
1356.20
|
14.1008
|
These 3 modes are of medium intensity and are due to the bending motion of the entire molecule. 3 degenerate modes are produced due to 3 different bending orientations
|
| 2
|
1356.20
|
14.1008
|
| 3
|
1356.20
|
14.1008
|
| 4
|
1578.58
|
0.0000
|
Modes 4 and 5 are degenerate symmetric bend modes while mode 6 is due to symmetric stretching. As there are no changes in dipole moment in these 3 modes, infrared is not absorbed, hence resulting in zero intensity.
|
| 5
|
1578.58
|
0.0000
|
| 6
|
3046.46
|
0.0000
|
| 7
|
3162.33
|
25.3343
|
3 degenerate stretching modes due to 3 different ways of stretching with degenerate energy levels. These 3 modes occur at higher energy level and intensity as stretching is more energetic than bending.
|
| 8
|
3162.33
|
25.3343
|
| 9
|
3162.33
|
25.3343
|
Charge Distribution
| Charge Distribution
|

|
Since the carbon atom in the center is more electronegative than the surrounding hydrogen atoms, it is not surprising that more electrons are concentrated towards the carbon center. Also, since all the C-H bonds are equal, it is not surprising that all 4 hydrogen atoms have the same charge.
|
Molecular Orbitals
| Molecular Orbitals
|
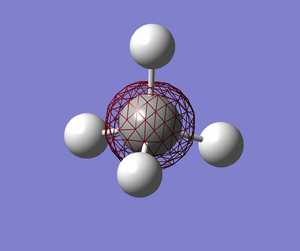
|
MO1 (E=-10.16707). This is a very low energy MO that is occupied. This MO is completely around the carbon atom only and it is probably due to the carbon atom's 1s atomic orbital (AO) only. In other words, this MO does not contribute to bonding. It is a non-bonding MO.
|
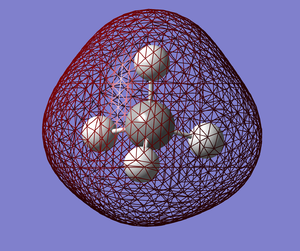
|
MO2 (E=-0.69041). This is a low energy bonding MO that is occupied. This MO completely surrounds the entire molecule and could possibly be due to the carbon atom's 2s atomic orbital (AO) and the 4 hydrogen atoms's 1s atomic orbitals (AOs) all being in phase.
|

|
MO3 (E=-0.38831) This is a bonding HOMO (Highest Occupied MO). This MO (MO3), MO4 and MO5 are degenerate. The only difference between these 3 degenerate MOs is their spatial orientations. This MO could possibly be due to 2 hydrogen atom's 1s atomic orbital being anti-phase to the other 2 hydrogen atom's 1s atomic orbital, while the carbon atom's 2p orbital is in phase with all 4 1s orbitals. There is only one nodal region and it is small, resulting in its low energy. Furthermore, the nodal region passes right through the center of the carbon atom, hence it does not affect the bond as it is not in between the carbon and hydrogen nuclei.
|

|
MO6 (E=+0.11824) This is an anti-bonding LUMO (Lowest Unoccupied MO). This MO looks similar to MO2 (from the outside) but it is larger than MO2 and it contains nodal regions as well. This MO could possibly be due to the carbon atom's 2s atomic orbital being anti-phase with all 4 hydrogen atoms's 1s atomic orbital. Due to its large size and nodal regions, this MO has a high energy.
|

|
MO7 (E=+0.17677) This is a high energy anti-bonding MO that is unoccupied. This MO (MO7), MO8 and MO9 are degenerate. This MO could possibly be due to 2 hydrogen atom's 1s atomic orbital being anti-phase to the other 2 hydrogen atom's 1s atomic orbital (just like in MO3), however, the carbon atom's 2p orbital is anti-phase with all 4 1s orbitals instead this time. The only difference between these 3 degenerate MOs is their spatial orientations. MO7 (MO8 and MO9 as well) is higher in energy compared to MO6, possibly due to more nodal regions.
|
H2O Molecule
| Information
|
|
|
| H2O Molecule
|
| Calculation Details
|
| Method
|
Basis Set
|
Point Group
|
| RB3LYP
|
6-31G(d,p)
|
C2v
|
| Calculation Results
|
| E(RB3LYP) /a.u
|
RMS Gradient /a.u.
|
| -76.41973740
|
0.00006276
|
| Click here for the log file.
|
(H2O) Item Value Threshold Converged?
Maximum Force 0.000099 0.000450 YES
RMS Force 0.000081 0.000300 YES
Maximum Displacement 0.000128 0.001800 YES
RMS Displacement 0.000120 0.001200 YES

















