
Rep:Mod:wcc14geeTS
Transition States and Reactivity
Introduction
- In this computational lab, three different pericyclic reactions are studied. They are Diels-Alder reaction (exercise 1), inverse electron demand Diels-Alder reaction (exercise 2) and Hetero Diels-Alder reaction (3). Diels-Alder reaction usually undergoes two pathways, endo and exo. However, competing pathway can exist like cheletropic. All these pathways can be analysed by the computational method in order to understand their rate and product stability.
- Gaussian is used to do all the calculations, the semi-empirical PM6 and DFT-B3LYP/6-31G(d) electronic structure methods are mainly used. The reactants and products of all reactions are optimised to the minimum and also all the transition states. PM6 is used to generate initial geometries while DFT-B3LYP gives a more detailed and accurate optimisation. This is because a higher number of the basis set is involved in the DFT-B3LYP calculation. A basis set is a set of functions that resemble the atomic orbitals (building block of molecular orbitals) and molecular orbitals are generated when basis sets combine linearly.
- Molecular vibrations are shown by performing frequency calculations, which can be used to confirm the position on the potential energy surface. A potential energy surface is a mathematical function that gives the relationship between the energy of the molecule and its geometry.[1] When all the vibrational frequencies are real, the structure is a minimum. The minima give the equilibrium position of the molecules, where the gradient is zero and the second derivative is positive and energy increases in both directions from this point. Minima usually correspond to the reactants and products. The minimum energy pathway between two minima is represented by the intrinsic reaction coordinate. When one imaginary vibrational frequency presence, the structure is at its transition state. The transition state is at the maximum of the minimum energy pathway with zero gradient and negative second derivative, energy decreases from this point. IRC calculations are calculated by force constants in order to predict the reaction path after transition state.
Nf710 (talk) 19:44, 22 February 2018 (UTC) This is corect but you have not shown understanding of these concepts on a 3N-6 dimensions PES and you have no discussed the computational methods either.
Exercise 1: Reaction of Butadiene with Ethylene
- A typical [4+2] cycloaddition Diels-Alder reaction is studied in this exercise. Reacting butadiene and ethylene forms cyclohexene by a concerted mechanism with a cyclic transition state. The reactants, transition state and product are optimised at semi-empirical PM6 level.
- The reaction scheme is show below.
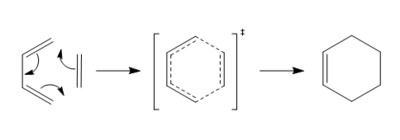
Figure 1: Reaction Scheme of Butadiene And Ethylene

Molecular Orbital Analysis
(Fv611 (talk) Your MO combinations are correct, but you have not considered the energies of your computed TS MOs: as this is a transition state, the energy of this structure will be higher than that of the product, and this will be reflected in the relative energies of the transition state MOs in the diagram.)
- The formation of the transition state is through the corresponding π molecular orbital interactions between the HOMO and LUMO of butadiene and ethylene. This is shown by Figure 2 and all the orbitals are labelled with their symmetry (S=symmetric and AS=antisymmetric). The formation of the transition state can be visualised by the JMol objects of the reactants (HOMO and LUMO of butadiene and ethylene) and the transition state orbitals (2π, 3π, 4π* and 5π*).
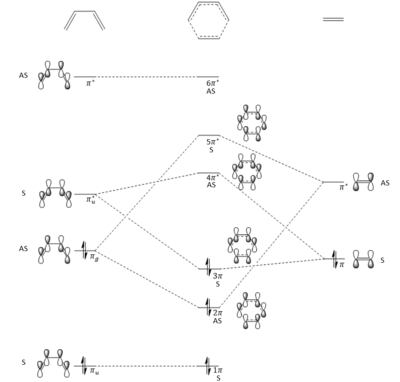
Figure 2: Molecular Orbital of The Diels-Alder Reaction between Butadiene and Ethylene - The MO theory states that a reaction is allowed when the molecular orbitals with same symmetry interact with each other, meaning symmetric-symmetric interaction and antisymmetric-antisymmetric interaction. Molecular orbitals with different symmetry cannot interact with each other, therefore symmetric-antisymmetric interaction is forbidden. In order to overlap efficiently, the molecular orbitals should be close in energy.
- In this reaction, the antisymmetric molecular orbital of butadiene (HOMO) interacts with the antisymmetric molecular orbital of diethylene (LUMO) to give the 2π and 5π* molecular orbitals of the transition state; the symmetric molecular orbital of butadiene (LUMO) interacts with the symmetric molecular orbital of diethylene (HOMO) to give the 3π and 4π* molecular orbitals of the transition state.
- The overlap integral is zero in a symmetric-asymmetric interaction while non-zero in symmetric-symmetric and antisymmetric-antisymmetric interactions.

Bond Length Analysis
- As the reaction progress, the bond length changes. Therefore, the bond lengths between different carbon atoms for reactants, transition state and product are studied. The position of different carbon atoms and the result is shown in Figure 3 and Table 1 respectively.
 |
|
|||||||||||||||||||||||||||||||||||||||
Table 2: Literature Values of C-C Bond Lengths And Van der Waals Radius of Carbon Atom Csp3-Csp3 Csp2-Csp2 Van der Waals Radius of C atom Bond Length / Å 1.54 [2] 1.33 [2] 1.77 [3]
- As seen from the results, during the reaction the reactants' double bonds ( C1-C2, C3-C4 and C5-C6) increase from 1.33Å to 1.54Å. This means the bonds are changing from Csp2-Csp2 to Csp3-Csp3 as the value matches with the literature (Table 2). On the other hand, C4-C5 decreases from 1.47Å to 1.34Å, indicating the formation of C=C (sp2) at the product. The new bonds formed at C2-C3 and C6-C1 are single bond (sp3) with bond length around 1.54Å.
- At the transition state, the distance between C2-C3 and C6-C1 is 2.11Å. Since the distance is shorter than twice the Van der Waals radii of carbon (3.4Å), therefore a bond is forming between C2-C3 and C6-C1.
Vibration Analysis
- As mentioned above, this [4+2] cycloaddition reaction undergoes a concerted mechanism. A concerted mechanism means all the bond breaking and forming takes place in parallel.[4]
Table 3: IRC and Negative Frequency at The Transition State IRC Negative Frequency (-948) at The Transition State 


- The IRC and the negative frequency at the transition state proves that this is a concerted reaction since both carbons on the ethylene approach butadiene's carbons at the same time. Therefore, the formation of the two new bonds are synchronous.
Exercise 2: Reaction of Cyclohexadiene and 1,3-Dioxole
- In this section, another Diels-Alder reaction is studied. The reaction between cyclohexadiene and 1,3-dioxole can go under two pathways (endo and exo) to form two products. This is shown in the reaction scheme below. The reaction barrier and reaction energy of the two pathways are calculated by Gaussian in order to determine the kinetic and thermodynamically products. All reactants, transition states and products are optimised to DFT B-3LYP/6-31G(d) level.

Figure 4: Reaction Scheme for The Reaction between Cyclohexadiene and 1,3-Dioxole

Molecular Orbital Analysis
(Fv611 (talk) As in the previous exercise, your combinations are correct but the relative energies of your TS MOs are not. Additionally you do not discuss at all the differences between the MO diagrams of the endo and of the exo conformation.)
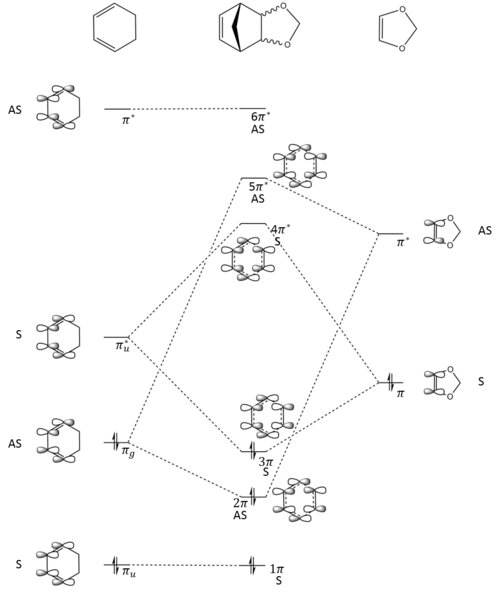
Figure 5: Reaction Scheme for The Reaction between Cyclohexadiene and 1,3-Dioxole

- The MO diagram for this reaction is shown in Figure 5. As mentioned in exercise 1, only molecular orbitals with same symmetry can interact. From both the Jmol objects of endo and exo transition state, it can be seen that both HOMO and LUMO of the transition states are symmetric. Referring to the MO diagram, this means the LUMO of cyclohexadiene interacts with the HOMO of 1,3-dioxole to produce the 3π (HOMO) and 4π* (LUMO) orbitals of the transition states. This is confirmed by calculating the energy of the orbitals for both transition states using single point energy calculation.
- This also suggests an inverse electron demand Diels-Alder reaction, cycloaddition between an electron-rich dienophile and an electron-poor diene. The energy level of the dienophile is raised by the electron donating oxygens in 1,3-dioxole leading to LUMOcyclohexadiene and HOMO1,3-dioxole are closer in energy than the HOMOcyclohexadiene and LUMO1,3-dioxole. Thus LUMOcyclohexadiene and HOMO1,3-dioxole are the frontier orbitals that interact the stongest and hence a shift of MO energy order.[5] [6]
Nf710 (talk) 19:50, 22 February 2018 (UTC) You can quantitively investigate this by running a single point energy calc on the reactant geoms on the irc.
Activation and Reaction Energies Analysis
- Table 4 shows the energy of reactants, products and transition states calculated by Gaussian. The energy of the reactants is the sum of the energies of cyclohexadiene and 1,3-dioxole, assuming that they are separated and have no interaction at the very beginning of the reaction. A reaction profile is drawn using these data.
|
 | |||||||||||||||||||||||||||
- In a Diels-Alder reaction, the endo pathway is typically kinetically favourable. This is reflected in the calculated results since the endo pathway has a lower activation energy (Figure 6) and a more stable transition state. This is due to the secondary orbital interaction between the oxygen and p-orbitals of cyclohexadiene (Figure 7).[7] Secondary orbital interaction is not expected in the exo pathway because the oxygens are pointed away from the cyclohexadiene p-orbitals.
 |
 |
- In a typical Diels-Alder reaction, the Exo product is expected to be more thermodynamically favourable because of less sterically crowded. However, the calculated results show that the endo product is more thermodynamically favourable. This can be explained by the possible steric clash between bridging and terminal hydrogens in the Exo product (Figure 8). The steric clash in the exo product raises the energy and hence leading the endo product being both kinetically and thermodynamically favourable.
Nf710 (talk) 19:53, 22 February 2018 (UTC) your energies are correct and you have come to the correct conclusion. And you have some nice diagrams which aid to the understanding of what you are saying. You could have gone into a bit more detail at some points such as investigating the electron demand of the reaction qualitatively. but other than that this was a good section.
Exercise 3: Diels-Alder vs Cheletropic
- In this exercise, the reaction between o-xylylene and sulphur dioxide is studied. Sulphur dioxide acts as the dienophile and reacts with o-xylylene via Hetero-Diels-Alder reaction with both endo and exo 6-membered ring products being formed. They can also react through cheletropic forming a 5-membered ring. The reaction scheme is shown below. With the semi-empirical PM6 methods all reactants, transition states and products are optimised. Reaction barriers and reaction energies are used to determine which reaction pathway is the most favourable.

Figure 9: Reaction Scheme for O-Xylylene And Sulphur Dioxide

Intrinsic Reaction Coordinate Calculations Analysis
- IRC calculations are done in both directions from the transition state using the semi-empirical PM6 method. The results from the three different reaction pathways are summarised below.
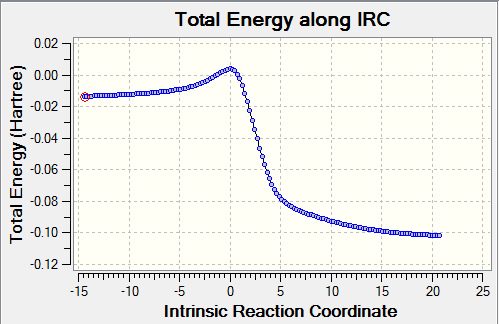
Table 5: IRC Caluclations and Total Energy along IRC for Different Pathways Pathway IRC Total Energy along IRC Diels-Alder Reaction via Endo 
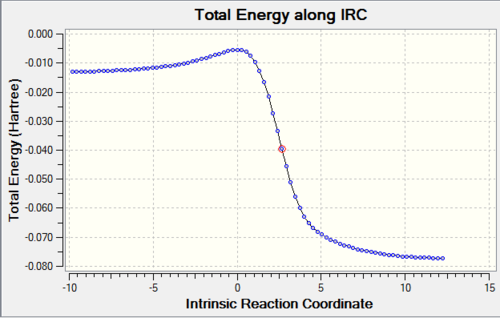
Diels-Alder Reaction via Exo 
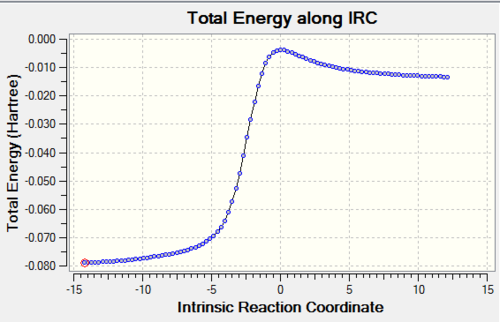
Cheletropic Reaction 






Activation and Reaction Energies Analysis
- The energy of reactants, products and transition states are calculated by Gaussian and shown in Table 6. The energy of the reactants is the sum of the energies of o-xylylene and sulphur dioxide assuming that they are separated and have no interaction at the very beginning of the reaction. A reaction profile is produced using these data.
|
 | |||||||||||||||||||||||||||||||||
(Your reactants are a bit too high in energy Tam10 (talk) 15:13, 26 February 2018 (UTC))
- From the reaction profile, the cheletropic product is the most stable and hence the cheletropic pathway is more thermodynamically favourable. This is because of the presence of the two strong S=O bonds and aromaticity.
- Diels-Alder reactions are more kinetically favoured compared to the cheletropic reaction as they have lower activation energies. Similar to the reaction in exercise 2, the endo transition state is more stable than the exo transition state as the endo approach has secondary orbital interactions between the oxygens and o-xylylene p-orbitals. Due to the greater steric clash within the endo product, the exo product is more stable.
Instability of o-Xylylene
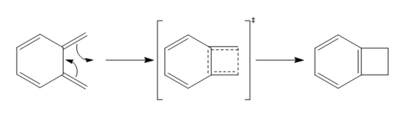
Figure 11: Electrocyclic Reaction of o-Xylylene

- o-Xylylene is not following the Hückel rule[8] (4n +2 electrons) so it does not have stabilisation from aromaticity, thus highly unstable. During the reaction, the 6-membered ring of o-xylylene aromatises to form a benzene ring, hence the products gain stability from aromaticity and are thermodynamically more stable than the reactants.
- The formation of benzocylobutane through electrocyclic reaction is another reaction pathway that can happen apart from Diels-Alder and cheletropic reaction.
- The driving force for these reactions (Diels-Alder, cheletropic and electrocyclic) is the formation of the aromatic benzene ring, stabilising the reaction to become more thermodynamically favourable.
(This is true, but have you calculated the activation energy for this reaction to say that it really is in competition? Tam10 (talk) 15:13, 26 February 2018 (UTC))
Side Reaction Analysis
- The internal cis-butadiene fragment in o-xylylene can also undergo Diels-Alder reaction forming both endo and exo products. The reaction scheme is shown below with the internal cis-butadiene fragment in light purple.
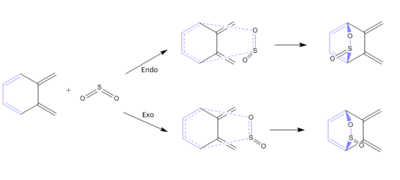
Figure 12: Diels-Alder Reaction between Internal Butadiene Fragment of o-Xylylene And Sulphur Dioxide

- IRC calculations are shown below.
| Pathway | IRC | Total Energy along IRC |
|---|---|---|
| Endo |  |
 |
| Exo |  |
 |
- Similar to the other reactions, the energies of the reactants, transition states and products are calculated and summarised in Table 7.
| Pathway | Energy / kJ mol-1 | ||||
|---|---|---|---|---|---|
| Reactants | Transition States | Products | Activation Energy | Reaction Energy | |
| Endo | 157.92 | 267.98 | 172.26 | 110.06 | 14.34 |
| Exo | 157.92 | 275.82 | 176.71 | 117.90 | 18.78 |
- A rection profile containing reactions with sulphur dioxide reacting with both external and internal butadiene fragment of o-xylene is conducted.

Figure 13: Reaction Profile for Diels-Alder Reaction between External and Internal Butadiene Fragment of o-Xylylene And Sulphur Dioxide

- As seen from Figure 13, reactions with internal cyclobutadiene fragments are both kinetically and thermodynamically unfavourable. Also, reactions reacting with the internal butadiene fragment, both endo and exo transition states have higher activation energy compared with previous reactions. The reaction energy suggested that reactions with internal butadiene fragment are endothermic.
Conclusion
- Three different pericyclic reactions are studied in this computational lab. The reactants, transition states and products of these reactions are optimised with the semi-empirical PM6 or DFT-B3LYP/6-31G(d) methods. Through Gaussian calculation, energies, vibrational frequencies and intrinsic reaction coordinates for all the molecules are obtained.
- In exercise 1, a [4+2] cycloaddition reaction between butadiene and ethylene is studied. It can be concluded that in order for molecular orbitals to interact, they have to have the same symmetry (symmetric and symmetric interaction or antisymmetric and antisymmetric interaction). Reactions with different symmetry (symmetric and antisymmetric interaction) is forbidden. The negative vibrational frequency of the transition state and IRC proved that this reaction proceeds via a concerted mechanism meaning the formation of new bonds are synchronous.
- Another Diels-Alder reaction is studied in exercise 2. The reaction between cyclohexadiene and 1,3-dioxole can proceed via two pathways, endo and exo. This is an inverse electron demand Diels-Alder reaction meaning the LUMOcyclohexadiene and HOMO1,3-dioxole are the frontier orbitals and interact the strongest. The calculated activation energy shows that the endo pathway is more kinetically favourable because of secondary orbital interaction between the oxygens and the p-orbital of cyclohexadiene. Since steric hindrance is present in the exo product, therefore raises its energy leading the endo product being thermodynamic. Thus, the endo pathway in this reaction is both kinetically and thermodynamically favourable.
- The reaction between o-xylylene and sulphur dioxide is the final reaction. This reaction can undergo via Diels-Alder or cheletropic. Similar to the reaction in exercise 2, if this reaction proceeds via Diels-Alder, it can form both endo and exo products. Once again, the endo pathway has the lowest activation energy hence most kinetically favourable. The cheletropic product is the thermodynamically favourable product because it is the most stable. The driving force for these reactions is the formation of the aromatic benzene ring, stabilising the reactions to become more thermodynamically favourable. When sulphur dioxide reacts with the internal butadiene fragment of o-xylylene, both endo and exo Diels-Alder reactions are endothermic and thermodynamically and kinetically unfavourable.
References
- ↑ Potential Energy Surface, http://vergil.chemistry.gatech.edu/courses/chem6485/pdf/pes-lecture.pdf, (accessed Feb 2018)
- ↑ 2.0 2.1 A. G. Orpen, L. Brammer, F. H. Allen, O. Kennard, D. G. Watson and R. Taylor, J. Chem. Soc. Dalt. Trans., 1987, S1–S83.
- ↑ A. Bondi, J. Phys. Chem., 1964, 68, 441–451.
- ↑ M. J. S. Dewar, J. J. P. Stewart and S. Olivella, J. Am. Chem. Soc., 1986, 108, 5771–5779.
- ↑ P. Rooshenas, K. Hof, P. R. Schreiner and C. M. Williams, European J. Org. Chem., 2011, 2011, 983–992.
- ↑ A. T. Dang, D. O. Miller, L. N. Dawe and G. J. Bodwell, Org. Lett., 2008, 10, 233–236.
- ↑ Secondary Orbital Interaction, http://www.cpp.edu/~psbeauchamp/pdf/316_MO_DA_rxn.pdf, (accessed Feb 2018)
- ↑ W. E. von Doering and F. L. Detert, J. Am. Chem. Soc., 1951, 73, 876–877.