
Rep:Mod:ssn3617
NH3 molecule
Optimization
Optimization was done using B3LYP method with 6-31G(d,p) basis set.
Final energy E(RB3LYP) = -56.55776873 a.u.
RMS gradient = 0.00000485 a.u.
Point group = C3V
Optimized N-H bond distance = 1.01798Å
Optimized H-N-H bond angle = 105.741°
Checking output summary file, it was confirmed that the final set of forces and displacements have converged, such that external forces are at a minimum.
"Item" table of converged forces and distances for this optimization
Item Value Threshold Converged? Maximum Force 0.000004 0.000450 YES RMS Force 0.000004 0.000300 YES Maximum Displacement 0.000072 0.001800 YES RMS Displacement 0.000035 0.001200 YES
NH3 molecule |
Optimization log file: here
Frequency Analysis

Expected number of modes = 3(4)-6 = 6 using 3N-6 rule for non-linear molecules (NH3 is trigonal pyramidal)
Modes 2 and 3 are degenerate, and modes 5 and 6 are degenerate.
'Bending' vibrations: Modes 1, 2, 3
'Stretching' vibrations: Modes 4, 5, 6
Mode 4 is highly symmetric, and mode 1 is known as the 'umbrella' mode
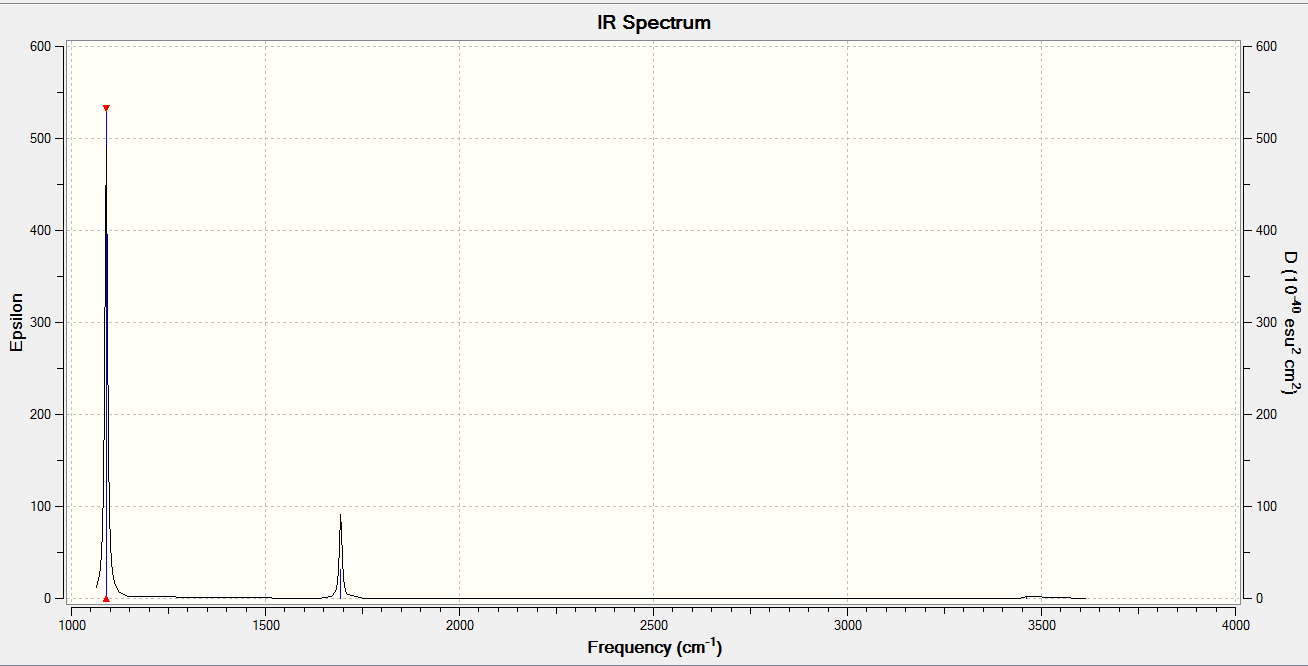
A total of 2 bands are expected to be seen in an experimental spectrum of gaseous ammonia as the stretching modes result in no(/minimal) net change in dipole and so the intensity of the peaks in an experimental IR spectrum would be too weak to be identified and separated from noise in the spectrum. We do not observe 3 bands despite there being 3 bending modes as modes 2 and 3 are degenerate, both occurring at the same frequency of 1693.95cm-1.
To check that this is true, click here for the spectrum.
{kind=link}
Charge Analysis
Charge on N-atom = -1.125
Charge on H-atoms = +0.375
N (χP = 3.04) is more electronegative than H (χP = 2.20). As such, we expect that in a molecule NH3, N would be more able to attract bonding electrons (in N-H bonds) to itself and hence have a more negative charge. This is in agreement with the results calculated from the optimization done on the NH3 molecule.
N2 molecule
Optimization
Optimization was done using B3LYP method with 6-31G(d,p) basis set.
Final energy E(RB3LYP) = -109.52412868 a.u.
RMS gradient = 0.00000060 a.u.
Point group = D∞h
Optimized N-N bond distance = 1.10550Å
Optimized N-N bond angle = 180°
Checking output summary file, it was confirmed that the final set of forces and displacements have converged, such that external forces are at a minimum.
"Item" table of converged forces and distances for this optimization
Item Value Threshold Converged? Maximum Force 0.000001 0.000450 YES RMS Force 0.000001 0.000300 YES Maximum Displacement 0.000000 0.001800 YES RMS Displacement 0.000000 0.001200 YES
N2 molecule |
Optimization log file: here
Frequency Analysis
Expected number of modes = 3(2)-5 = 1 using 3N-5 rule for linear molecules (N2 is linear)
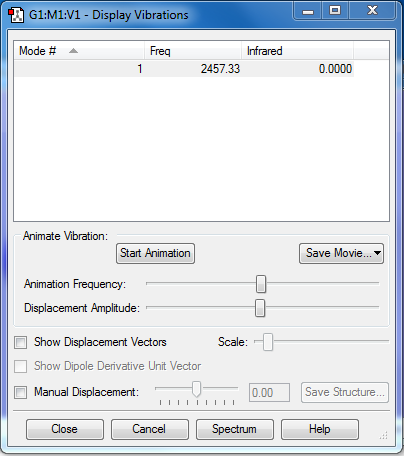
There is only 1 vibrational mode occurring at 2457.33cm-1 and it is not negative, as seen here.
{kind=link}
H2 molecule
Optimization
Optimization was done using B3LYP method with 6-31G(d,p) basis set.
Final energy E(RB3LYP) = -1.17853936 a.u.
RMS gradient = 0.00000017 a.u.
Point group = D∞h
Optimized H-H bond distance = 0.74279Å
Optimized H-H bond angle = 180°
Checking output summary file, it was confirmed that the final set of forces and displacements have converged, such that external forces are at a minimum.
"Item" table of converged forces and distances for this optimization
Item Value Threshold Converged? Maximum Force 0.000000 0.000450 YES RMS Force 0.000000 0.000300 YES Maximum Displacement 0.000000 0.001800 YES RMS Displacement 0.000001 0.001200 YES
H2 molecule |
Optimization log file: here
Frequency Analysis
Expected number of modes = 3(2)-5 = 1 using 3N-5 rule for linear molecules (H2 is linear)
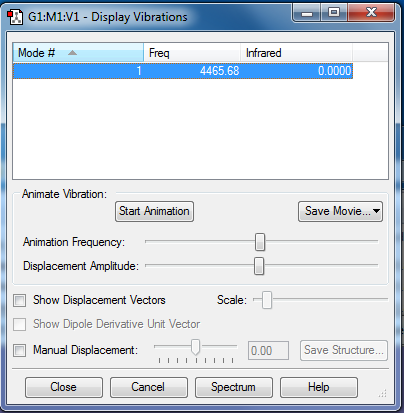
There is only 1 vibrational mode occurring at 4465.68cm-1 and it is not negative, as seen here.
{kind=link}
Haber-Bosch process
E(NH3)= -56.55776873 a.u.
2*E(NH3)= -113.1155375 a.u.
E(N2)= -109.52412868 a.u.
E(H2)= -1.17853936 a.u.
3*E(H2)= -3.53561808 a.u.
ΔE=2*E(NH3)-[E(N2)+3*E(H2)]= -0.0557907 a.u. = -146.48 kJ/mol (2 d.p.)
Hence, the amount of energy required to convert hydrogen and nitrogen gas into ammonia gas is -146.48 kJ/mol. Since this reaction is exothermic, we can deduce that the ammonia product is more stable as energy is released upon its formation.
Project molecule: O2
Optimization
Optimization was done using B3LYP method with 6-31G(d,p) basis set.
Final energy E(RB3LYP) = -150.25742434 a.u.
RMS gradient = 0.00007502 a.u.
Point group = D∞H
Optimized O=O bond distance = 1.21602Å
Optimized O=O bond angle = 180°
Checking output summary file, it was confirmed that the final set of forces and displacements have converged, such that external forces are at a minimum.
"Item" table of converged forces and distances for this optimization
Item Value Threshold Converged? Maximum Force 0.000130 0.000450 YES RMS Force 0.000130 0.000300 YES Maximum Displacement 0.000080 0.001800 YES RMS Displacement 0.000113 0.001200 YES
O2 molecule |
Optimization log file: here
Frequency Analysis

Expected number of modes = 3(2)-5 = 1 using 3N-5 rule for linear molecules (O2 is linear)
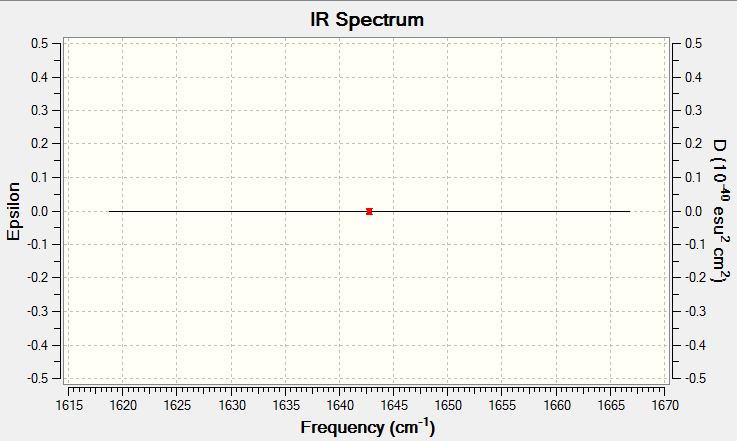
There is a single symmetrical 'stretching' vibrational mode occurring at 1642.74cm-1.
No bands would be expected to be seen in an experimental IR spectrum as the stretch is symmetric and the molecule is non-polar. As such, there would be no net change in dipole moment, giving no peaks.
To verify, click here.
{kind=link}
Charge Analysis
There are is no charge on both O atoms.
Because the O atoms have the same electronegativity, χP = 3.44, we expect that in a molecule O2, both atoms will be equally able to attract bonding electrons (in O=O bond) to themselves. Since the bonding electrons are equally shared by the two atoms, there will be no charge on the O atoms. This is in agreement with the results calculated from the optimization done on the HCl molecule.
Molecular Orbitals
Here is an image showing the energy levels of the O2 MOs to be discussed below.
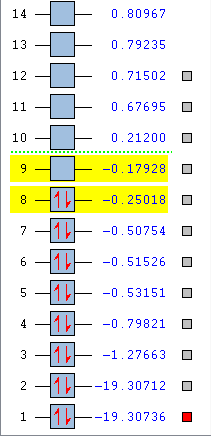{kind=link}
1: 
This shows the MO deepest in energy at -19.30736 a.u.
There is no overlap between the AOs on the O atoms and hence this is a non-bonding MO, occupied by the own electrons of each O atom. The AOs are in phase in this MO. These electrons are held tightly to the respective O atoms and are not involved in chemical reactions.
2: 
This is the MO that is next deepest in energy at a slightly higher energy level of -19.30712 a.u.
As can be seen by its energy level, this MO is still very deep in energy. There is no overlap between the AOs on the O atoms and hence this is a non-bonding MO, occupied by the own electrons of each O atom. The AOs are in phase in this MO. These electrons are held tightly to the respective O atoms and are not involved in chemical reactions.
3: 
This MO has an energy level of -1.27663 a.u.
This is the MO resulting from the overlap of the 2s AOs of the O atoms, and it is the resulting bonding MO since it is the 3rd deepest in energy, following the two above MOs. This is a σg MO which is occupied. The electrons in this MO are involved in chemical reactions.
4: 
This is the MO 5th deepest in energy with energy -0.53151 a.u.
There is no s-p mixing that needs to be considered in the case of O2 as the energy gap between the 2s and 2p AOs are sufficiently large such that the effect of s-p mixing can effectively be ignored.
This MO is the result of the head-on overlap of two 2p AOs of the O atoms, and it is a bonding MO.
This is a σg MO which is occupied, and the electrons in this MO are involved in chemical reactions.
5: 
This is the MO 8th deepest in energy with energy -0.25018 a.u.
This MO results from the sideways overlap of two 2p AOs of the O atoms and is an anti-bonding orbital. This πg* MO is in the HOMO/LUMO region. The MO is occupied and is specifically the HOMO, and as such electrons in this MO are often involved in chemical reactions.
6: 
This is the MO 9th deepest in energy with energy -0.17928 a.u.
This MO results from the sideways overlap of two 2p AOs of the O atoms and is an anti-bonding orbital. This πg* MO is in the HOMO/LUMO region. The MO is unoccupied and is specifically the LUMO, and as such is involved in chemical reactions, specifically used to accommodate electrons from reacting species.
Extension: HCl
Optimization
Optimization was done using B3LYP method with 6-31G(d,p) basis set.
Final energy E(RB3LYP) = -460.80077875 a.u.
RMS gradient = 0.00005211 a.u.
Point group = C∞v
Optimized H-Cl bond distance = 1.28599Å
Optimized H-Cl bond angle = 180°
Checking output summary file, it was confirmed that the final set of forces and displacements have converged, such that external forces are at a minimum.
"Item" table of converged forces and distances for this optimization
Item Value Threshold Converged? Maximum Force 0.000090 0.000450 YES RMS Force 0.000090 0.000300 YES Maximum Displacement 0.000139 0.001800 YES RMS Displacement 0.000197 0.001200 YES
HCl molecule |
Optimization log file: here
Frequency Analysis

Expected number of modes = 3(2)-5 = 1 using 3N-5 rule for linear molecules (HCl is linear)
There is a single 'stretching' vibrational mode occurring at 2956.80cm-1.
A total of 1 band would be expected to be seen in an experimental IR spectrum.
To verify, click here.
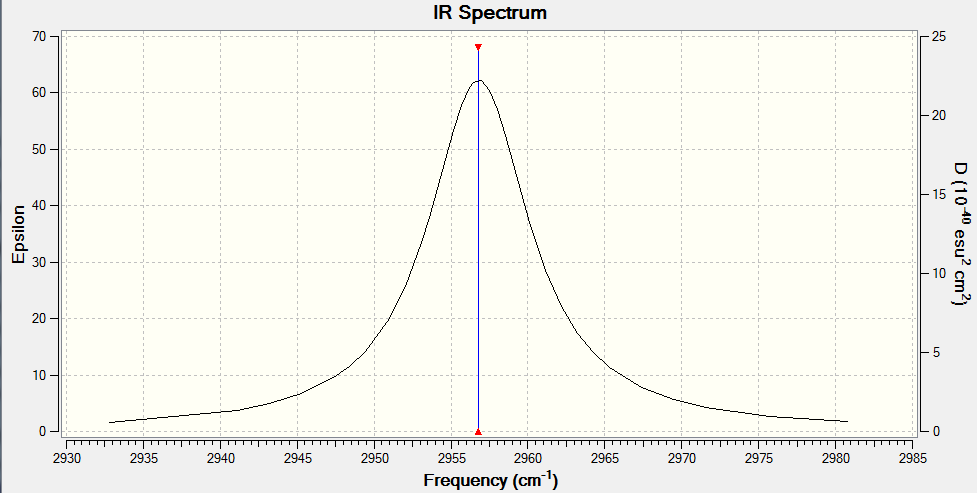{kind=link}
Charge Analysis
Charge on Cl atom = -0.284
Charge on H atoms = +0.284
Cl (χP = 3.16) is more electronegative than H (χP = 2.20). As such, we expect that in a molecule HCl, Cl would be more able to attract bonding electrons (in H-Cl bond) to itself and hence have a more negative charge. This is in agreement with the results calculated from the optimization done on the HCl molecule.
Molecular Orbitals
Here is an image showing the energy levels of the HCl MOs to be discussed below.
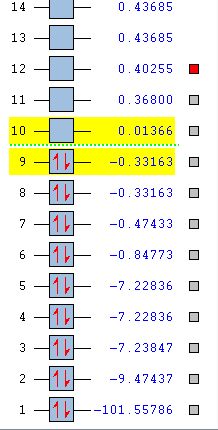{kind=link}
Both HCl and O2 are linear, but we expect some differences in their MOs as HCl is polar while O2 is non-polar. We will investigate this below.
1: 
The MO deepest in energy is at -101.55786 a.u.
This is a non-bonding σ MO that stems from the 1s AO of the Cl atom which is very deep in energy. Only electrons of Cl occupy this MO and they do not participate in chemical reactions, but rather stay bonded to the Cl atom.
2: 
This is the MO second deepest in energy at -9.47437 a.u.
This is the non-bonding σ MO that can be attributed to the 2s AO of the Cl atom. This MO is deep in energy and occupied only by the electrons of Cl. This is due to the highly electronegative nature of the Cl atom. Since they are tightly held to the Cl nuclei, they do not participate in chemical reactions, and stay bonded to the Cl atom. The 2s AO does not overlap with that of the 1s AO of the H atom as it is much lower in energy.
3: 
This is the MO 6th deepest in energy at -0.84773 a.u.
This is the bonding σg MO resulting from the overlap of the 3s AO of the Cl atom and the 1s AO of the H atom. The MO is occupied and electrons in the MO are involved in chemical reactions. It can also be observed that there is slightly more electron density about the Cl atom as compared to the H atom and this can be attributed to the higher electronegativity of Cl (χP = 3.16) compared to H (χP = 2.20) and as such, Cl atom is more able to attract electrons in the bond to itself as compared to H. This gives more electron density about Cl as compared to H.
4: 
This is the MO 7th deepest in energy at -0.47433 a.u.
This is the bonding σ MO resulting from the overlap of the 3pz AO of the Cl atom and the 1s AO of the H atom. The MO is occupied and since energy is near the HOMO region, electrons in the MO are involved in chemical reactions.
5: 

These are the MOs 8th and 9th deepest in energy at -0.33163 a.u. and they are degenerate.
They are non-bonding and formed from the 3px and 3py AOs of the Cl atom. There is no overlap with the AO of the H atom unlike in 3pz as the symmetry requirements are not met. The two non-bonding MOs are occupied and one can be considered as the HOMO, with the electrons in these MOs being involved in chemical reactions. The high energy level of the MOs allow for electrons in these MOs to be much more easily removed than those of MOs deeper in energy.
6: 
This is the MO 10th deepest in energy at +0.01366 a.u.
This is the anti-bonding σ* MO resulting from the overlap of the 3pz AO of the Cl atom and the 1s AO of the H atom. The MO is unoccupied and this is the LUMO of the molecule. The MO is involved in chemical reactions, accepting electrons from incoming reactants. It is the unoccupied MO of deepest energy and hence is the most stable MO that new electrons can be accommodated in.