
Rep:Mod:sk716
NH3 molecule
- Calculation method: RB3LYP;
- Basis set: 6-31G(d,p);
- Final energy = -56.55776873 au;
- RHS gradient = 0.00000485 au;
- Point group: C3v;
- N-H bond distance = 1.01798 Å;
- H-N-H bond angle = 105.741 degrees.
Item Value Threshold Converged?
Maximum Force 0.000004 0.000450 YES
RMS Force 0.000004 0.000300 YES
Maximum Displacement 0.000072 0.001800 YES
RMS Displacement 0.000035 0.001200 YES
Predicted change in Energy=-5.986295D-10
Optimization completed.
-- Stationary point found.
----------------------------
! Optimized Parameters !
! (Angstroms and Degrees) !
-------------------------- --------------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------------------
! R1 R(1,2) 1.018 -DE/DX = 0.0 !
! R2 R(1,3) 1.018 -DE/DX = 0.0 !
! R3 R(1,4) 1.018 -DE/DX = 0.0 !
! A1 A(2,1,3) 105.7412 -DE/DX = 0.0 !
! A2 A(2,1,4) 105.7412 -DE/DX = 0.0 !
! A3 A(3,1,4) 105.7412 -DE/DX = 0.0 !
! D1 D(2,1,4,3) -111.8571 -DE/DX = 0.0 !
--------------------------------------------------------------------------------
Optimised ammonia |

- Vibration modes = 6;
- Modes 2 & 3 and 5 & 6 are degenerate;
- Modes 2 & 3 are bending vibrations and modes 4, 5 & 6 are stretching vibrations;
- Modes 4 are highly symmetric;
- Mode 1 is the umbrella mode;
- 4 bands are expected to be observed in the spectrum;;
- Charge of N = -1.125;
- Charge of H = +0.375;
I'd expect N to have a negative charge and H to have a positive charge because nitrogen's more electronegative, so a lot of the electron density will be around the N atom;;
N2 molecule
- Calculation method: RB3LYP;
- Basis set: 6-31G(d,p);
- Final energy = -109.52412868 au;
- RHS gradient: 0.00000060 au;
- Point group: D∞h
- Bond length = 1.10550 Å;
Item Value Threshold Converged?
Maximum Force 0.000001 0.000450 YES
RMS Force 0.000001 0.000300 YES
Maximum Displacement 0.000000 0.001800 YES
RMS Displacement 0.000000 0.001200 YES
Predicted change in Energy=-3.401015D-13
Optimization completed.
-- Stationary point found.
----------------------------
! Optimized Parameters !
! (Angstroms and Degrees) !
-------------------------- --------------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------------------
! R1 R(1,2) 1.1055 -DE/DX = 0.0 !
--------------------------------------------------------------------------------
Optimised dinitrogen |

There are no negative frequencies
H2 molecule
- Calculation method: RB3LYP;
- Basis set: 6-31G(d,p);
- Final energy = -1.17853936 au;
- RHS gradient: 0.00000017 au;
- Point group: D∞h;
- Bond length = 0.74279 Å;
Item Value Threshold Converged?
Maximum Force 0.000000 0.000450 YES
RMS Force 0.000000 0.000300 YES
Maximum Displacement 0.000000 0.001800 YES
RMS Displacement 0.000001 0.001200 YES
Predicted change in Energy=-1.164080D-13
Optimization completed.
-- Stationary point found.
----------------------------
! Optimized Parameters !
! (Angstroms and Degrees) !
-------------------------- --------------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------------------
! R1 R(1,2) 0.7428 -DE/DX = 0.0 !
--------------------------------------------------------------------------------

Optimised dihydrogen |
E(NH3)= -56.55776873 au
2×E(NH3)= -113.1155375 au
E(N2)= -109.52412868 au
E(H2)= -1.17853936 au
3×E(H2)= -3.53561808 au
ΔE=2×E(NH3)-[E(N2)+3×E(H2)]= 0.05579074 au = -146.4785879 kJ mol-1
A literature value estimates the enthalpy change of reaction as -50 kJ mol-1[1], which is 3 times less than the prediction using the calculations above. This difference could be due to the calculation method as it uses purely theoretical methods to calculate the energy (which can be viewed as calculating the enthalpy change of reaction in a vacuum), whereas the experimental conditions are significantly different from this. The product is more stable than the reactants because the free energy is around negative 65 kJ mol-1[2]
H2CO analysis
- Calculation method: RB3LYP;
- Basis set: 6-31G(d,p);
- Final energy = -114.50319933 au;
- RHS gradient: 0.00007386 au;
- Point group: CS;
- C-H bond distance = 1.11057 Å;
- C=O bond distance = 1.20676 Å;
- H-C-O bond angle = 122.395 degrees.
Item Value Threshold Converged?
Maximum Force 0.000197 0.000450 YES
RMS Force 0.000085 0.000300 YES
Maximum Displacement 0.000252 0.001800 YES
RMS Displacement 0.000142 0.001200 YES
Predicted change in Energy=-3.772186D-08
Optimization completed.
-- Stationary point found.
----------------------------
! Optimized Parameters !
! (Angstroms and Degrees) !
-------------------------- --------------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------------------
! R1 R(1,2) 1.1106 -DE/DX = -0.0001 !
! R2 R(1,3) 1.1106 -DE/DX = -0.0001 !
! R3 R(1,4) 1.2068 -DE/DX = -0.0002 !
! A1 A(2,1,3) 115.2188 -DE/DX = 0.0 !
! A2 A(2,1,4) 122.3859 -DE/DX = 0.0 !
! A3 A(3,1,4) 122.3954 -DE/DX = 0.0 !
! D1 D(2,1,4,3) 180.0 -DE/DX = 0.0 !
--------------------------------------------------------------------------------
The molecule is fully optimised because the maximum force is less than 0.00045 au and the maximum displacement is less than 0.0003 au.
Formaldehyde molecule |
-
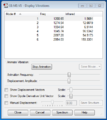 Vibration windows screenshot
Vibration windows screenshot -
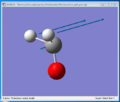 Symmetrical bending
Symmetrical bending -
 Symmetrical bending
Symmetrical bending -
 Symmetrical bending
Symmetrical bending -
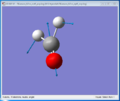 In-plane scissoring
In-plane scissoring -
 Symmetrical stretching
Symmetrical stretching -
 Asymmetrical stretching
Asymmetrical stretching







The values from the IR frequencies are a good estimate of the experimental values[3].

-
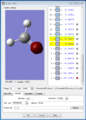 The 1s orbital of O (this is a non-bonding orbital because it's too deep in energy to overlap with other orbitals)
The 1s orbital of O (this is a non-bonding orbital because it's too deep in energy to overlap with other orbitals) -
 The 1s orbital of C (like O's 1s orbital, it's too deep in energy to overlap with other orbitals)
The 1s orbital of C (like O's 1s orbital, it's too deep in energy to overlap with other orbitals) -
 This can be viewed as the orbital made by the in-phase overlapping of the CH2 fragment and O's 2s orbital
This can be viewed as the orbital made by the in-phase overlapping of the CH2 fragment and O's 2s orbital -
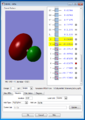 This is the antibonding molecular orbital for the interaction in the third image (O is in the opposite phase)
This is the antibonding molecular orbital for the interaction in the third image (O is in the opposite phase) -
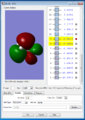 This is the LUMO of formaldehyde, which is the antibonding orbital of C's 2p orbital (which is orthogonal to the plane of the molecule, and O's 2p orbital (orthogonal as well). The energy of the MO is negative.
This is the LUMO of formaldehyde, which is the antibonding orbital of C's 2p orbital (which is orthogonal to the plane of the molecule, and O's 2p orbital (orthogonal as well). The energy of the MO is negative.




