
Rep:Mod:rup3110
Module 2
BH3

Optimised B-H bond length: 1.19435 angstrom
Optimised H-B-H angle: 120°
Results Summary from BH3 Optimisation
File Type: .log
Calulation Type: FOPT
Calculation Method: RB3LYP
Basis Set: 3-21G
Final Energy: -26.462 a.u.
Dipole Moment: 0.00 Debye
Point Group: D3H
Time Taken for Calculation: 25 seconds
Results from Vibrational Analysis of BH3

| No. | Form of vibration | Frequency | Intensity | Symmetry,
D3H point group |
| 1 |  |
1145.71 | 92.699 | A2’’ |
| 2 |  |
1204.66 | 12.379 | E’ |
| 3 |  |
1204.66 | 12.381 | E’ |
| 4 |  |
2592.79 | 0 | Totally symmetric,
A’1 |
| 5 |  One H atom moves in and out, all other atoms remain stationary. |
2731.31 | 103.837 | E’ |
| 6 |  |
2731.31 | 103.85 | E’ |
Although there are 6 different vibrations for BH3, there are only 5 peaks in the infrared spectrum. This is because a peak will only show in infrared spectroscopy if the is a change in dipole in the molecule. The 4th vibrational mode is totally symmetric so will not cause a change in dipole and hence will not appear in the infrared spectrum.
Results from Molecular Orbital Analysis for BH3

-
 BH3 Molececular Orbital 1a'1
BH3 Molececular Orbital 1a'1 -
 BH3 Molececular Orbital 2a'1-bonding
BH3 Molececular Orbital 2a'1-bonding -
 BH3 Molececular Orbital 1e'-bonding
BH3 Molececular Orbital 1e'-bonding -
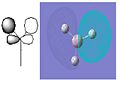 BH3 Molececular Orbital 2e' - HOMO-bonding
BH3 Molececular Orbital 2e' - HOMO-bonding -
 BH3 Molececular Orbital a2 - LUMO-non-bonding
BH3 Molececular Orbital a2 - LUMO-non-bonding -
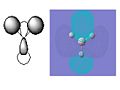 BH3 Molecular Orbital 3e'-antibonding
BH3 Molecular Orbital 3e'-antibonding -
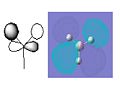 BH3 Molecular Orbital 4e'-antibonding
BH3 Molecular Orbital 4e'-antibonding -
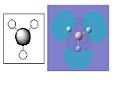 BH3 Molecular Orbital 3a'1-antibonding
BH3 Molecular Orbital 3a'1-antibonding








The pictures above show the comparison between the predicted molecular orbitals from the MO diagram and the computer generated orbitals from Gaussian. I have matched up the orbitals visually, the first orbital isn't on the diagram as it is much lower in energy and isn't really involved in the bonding of the molecule. The eneies of the orbitals roughly match to where they're representatives are located on the diagram except for the anti-bonding orbitals as the computer generated orbitals show that the 3a'1 is higher in energy than the 3 and 4e'.
BCl3

Optimised B-Cl bond length: 1.86592 angstrom
Optimised Cl-B-Cl bond angle: 120°
Results Summary from BCl3 Optimisation
File Type: .log
Calculation Type: FOPT
Calculation Method: RB3LYP
Basis Set: LANL2MB
Final Energy: -69.439 a.u.
Dipole Moment: 0.00 Debye
Point Group: D3H
Time Taken for Calculation: 12 seconds
SiCl4

Optimised Si-Cl bond length: 2.33723 angstrom (2.14045 a 3-21g)
Optimised Cl-Si-Cl bond angle: 109.471°
Results Summary from SiCl4 Optimisation
File Type: .log
Calculation Type: FOPT
Calculation Method: RB3LYP
Basis Set: LANL2MB (3-21g)
Final Energy: -63.655 a.u. (-2120.107 a.u.)
Dipole Moment: 0.00 Debye
Point Group: TD
Time Taken for Calcluation: 21 seconds (23s)
Co-ordinates:
Number Atomic Number Type X Y Z --------------------------------------------------------------------- 1 14 0 0.000000 0.000000 0.000000 2 17 0 1.349401 1.349401 1.349401 3 17 0 -1.349401 -1.349401 1.349401 4 17 0 -1.349401 1.349401 -1.349401 5 17 0 1.349401 -1.349401 -1.349401
NH3

Optimised N-H bond length: 1.00591 angstrom
Optimised H-N-H bond angle: 116.251°
Results Summary from NH3 Optimisation
File Type: .log
Calculation Type: FOPT
Calculation Method: RB3LYP
Basis Set: 6-31G
Overall Energy: -56.532 a.u.
Dipole Moment: 1.3238 Debye
Point Group: C3V
Time Taken for Calculation: 27 seconds
Results from Vibrational Analysis

| No. | Vibration | Frequency | Intensity |
| 1 |  |
452.302 | 599.472 |
| 2 |  |
1680.47 | 41.726 |
| 3 |  |
1680.47 | 41.425 |
| 4 |  |
3575.43 | 0.069 |
| 5 |  |
3775.76 | 7.089 |
| 6 |  |
3775.76 | 7.088 |
NH3 Distorted

Optimised N-H bond length: 1.00595 angstrom
Optimised H-N-H bond angle: 116.158°
Results Summary from NH3 Optimisation
File Type: .log
Calculation Type: FOPT
Calculation Method: RB3LYP
Basis Set: 6-31G
Overall Energy: -56.532 a.u.
Dipole Moment: 1.3383 Debye
Point Group: C1
Time Taken for Calculation: 42 seconds
NH3 High Symmetry

Optimised N-H bond length: 0.99807 angstrom
Optimised H-N-H bond angle: 120°
Results Summary from NH3 Optimisation
File Type: .log
Calculation Type: FOPT
Calculation Method: RMP2-FU
Basis Set: 6-311, G(d,p)
Overall Energy: -56.427 a.u.
Dipole Moment: 0.00 Debye
Point Group: D3H
Time Taken for Calculation: 26 seconds
Results from Vibrational Analysis

| No. | Vibration | Frequency | Intensity |
| 1 |  |
-840.58 | 487.811 |
| 2 |  |
1587.95 | 43.601 |
| 3 |  |
1587.95 | 43.601 |
| 4 |  |
3676.66 | 0 |
| 5 |  |
3894.23 | 60.032 |
| 6 |  |
3894.23 | 60.03 |
The diffrence in starting symmetry has made a difference in the resultins symmetry as the ordinary NH3 structure had ended up with C3V symmetry, the distorted NH3 structure has ended up with C1 symmetry and the high symmetry NH3 has ended up with D3H symmetry. The most symmetric starting molecule was the high symmetry NH3, which also took the least time to compute (26s). The least symmetric starting molcule was the distorted NH3 molecule, which took the lonest time to compute (46s). Being less symmetric means it is further away from the correct optimised structure,so it has to go throgh mre possibilites to reach the correct structure. This takes more time.
The molecules can break symmetry during optimisation, this is seen in the distorted molecule as it ends up very much lik the ordinary NH3.
It wouldn't be broken by a high symmetry moleculeas it is too close to a possible structure t deviate away, despite not necessarily being the lowest energy conformation.
The lowest energy geometry is that of the C3V and the distorted NH3 structure.
The energy difference between the D3H and C3V molecules is 275.68kJ/mol. This shows that the C3V structure is more stable than the D3H structre so is the favoured conformation.
The vibrational analysis shows 6 positive vibrational modes for the C3V molecule and 5 positive and 1 negative (-840.58) vibrational modes for the D3H molecule.
The first, second, third and forth vibrational modes for the 2 molecules have similar motion. The first follows the path of the inversion.
NH3
Optimised N-H bond length: 1.01287 angstrom
Optimised H-N-H bond angle: 107.453°
Results Summary from NH3 Optimisation
File Type: .log
Calculation Type: FOPT
Calculation Method: RMP2-FU
Basis Set: 6-311+G(d,p)
Overall Energy: -56.434 a.u.
Dipole Moment: 1.7874 Debye
Point Group: C3V
Time Taken for Calculation: 30 seconds
NH3 Distorted
Optimised N-H bond length: 1.01262 angstrom
Optimised H-N-H bond angle: 107.444°, 107.454, 107.448
Results Summary from NH3 Optimisation
File Type: .log
Calculation Type: FOPT
Calculation Method: RMP2-FU
Basis Set: 6-311, G(d,p)
Overall Energy: -56.434 a.u.
Dipole Moment: 1.7877 Debye
Point Group: C1
Time Taken for Calculation: 38 seconds
NH3 High Symmetry
Optimised N-H bond length: 0.99807 angstrom
Optimised H-N-H bond angle: 120°
Results Summary from NH3 Optimisation
File Type: .log
Calculation Type: FOPT
Calculation Method: RMP2-FU
Basis Set: 6-311, G(d,p)
Overall Energy: -56.427 a.u.
Dipole Moment: 0.00 Debye
Point Group: D3H
Time Taken for Calculation: 24 seconds
The NH3 optimisation took 3 seconds longer but the distorted NH3 took 4 seconds less and the high symmetry NH3 took 2 seconds less than the original calculations.
The energy barrier for inversion is 18.378kJ/mol. This is quite a bit smaller than the previous value from the other calculations, but is alot closr to the experimental value of 24.3kJ/mol, which shows that this calculation gives more accurate results for the geometries.
Isomers of Mo(CO)4L2
The following results are from calculations performed on Mo(CO)4(PCH3)3.
Trans Isomer
LANL2MB Optimisation
Mo-P bond distance: 2.647
Mo-C bond distance: 2.047
C-O bond distance: 1.204
P-C bond distance: 1.964
C-Mo-C bond angle: 90
P-Mo-P bond angle: 180
Calculation Type: FOPT
Calculation Method: RB3LYP
Basis Set: LANL2MB
Overall Energy: -764.475 a.u.
Dipole Moment: 0.012 Debye
Point Group: C1
Time Taken for Calculation: 1 hour 3 minutes 1.2 seconds
LANL2DZ Optimisation
Mo-P bond distance: 2.571
Mo-C bond distance: 2.028
C-O bond distance: 1.190
P-C bond distance: 1.890
C-Mo-C bond angle: 90
P-Mo-P bond angle: 180
Calculation Type: FOPT
Calculation Method: RB3LYP
Basis Set: LANL2DZ
Overall Energy: -773.357 a.u.
Dipole Moment: 0.008 Debye
Point Group: C1
Time Taken for Calculation: 2 hours 2 minutes 44.2 seconds
Vibrational Analysis

| Frequency | Intensity | Part of Molecule | ||
| 321.848 | 32.30 | P(CH3)3 | ||
| 428.529 | 36.43 | C=O | ||
| 589.942 | 58.39 | C=O | ||
| 635.644 | 22.03 | P(CH3)3 | ||
| 658.395 | 143.33 | C=O | ||
| 1018.02 | 390.51 | P(CH3)3 | ||
| 1367.62 | 59.21 | CH3 | ||
| 1499.7 | 39.13 | CH3 | ||
| 1840.92 | 2005.98 | C=O | ||
| 3056.55 | 84.87 | CH3 | ||
| 3162.35 | 82.66 | CH3 |
Cis Isomer

LANL2MB Optimisation
Mo-P bond disatnce: 2.745 (value from reference: 2.576)
Mo-C bond distance (axial): 2.051 (value from reference: 2.059)
Mo-C bond disatnce (equatorial): 1.991 value from reference: 1.972)
C-O bond distance (axial): 1.204 (value from reference: 1.136)
C-O bond distance (equatorial): 1.207 (vaule from reference: 1.158)
P-C bond distance: 1.968 (value from reference: 1.83-1.85)
C-Mo-C bond angle between axial: 180 (value from reference: 173)
C-Mo-C bond angle between equatorial: 90 (value from reference: 83)
P-Mo-P bond angle: 90 (value from reference: 104)
Calculation Type: FOPT
Calculation Method: RB3LYP
Basis Set: LANL2MB
Overall Energy: -764.484 a.u.
Dipole Moment: 7.450 Debye
Point Group: C1
Time Taken for Calculation: 44 minutes 7 seconds
Values for Mo(CO)4(PPh3)3 taken from "Steric contributions to the solid-state structures of bis(phosphine) derivatives of molybdenum carbonyl. X-ray structural studies of cis-Mo(CO)4[PPh3-nMen]2 (n = 0, 1, 2)", F. Albert Cotton, Donald J. Darensbourg, Simonetta Klein, and Brian W. S. Kolthammer, Inorg. Chem., 21, (1982), p294-299
LANL2DZ Optimisation
Mo-P bond distance: 2.648
Mo-C bond distance (axial): 2.033
Mo-C bond distance (equatorial): 1.982
C-O bond distance (axial): 1.188
C-O bond distance (equatorial): 1.192
P-C bond distance: 1.892
C-Mo-C bond angle between axial: 180
C-Mo-C bond angle between equatorial: 90
P-Mo-P bond angle: 90
Calculation Type: FOPT
Calculation Method: RB3LYP
Basis Set: LANL2DZ
Overall Energy: -773.360 a.u.
Dipole Moment: 9.072 Debye
Point Group: C1
Time Taken for Calculation: 10 hours 22 minutes 15.2 seconds
Vibrational Analysis

| Frequency | Intensity | Part of Molecule | ||
| 311.137 | 13.94 | P(CH3)3 | ||
| 421.271 | 43.40 | C=O | ||
| 589.664 | 58.77 | C=O | ||
| 606.533 | 59.00 | C=O | ||
| 634.045 | 65.91 | C=O | ||
| 1007.49 | 24.87 | P(CH3)3 | ||
| 1026.96 | 140.70 | P(CH3)3 | ||
| 1367.73 | 34.43 | CH3 | ||
| 1498.5 | 33.24 | CH3 | ||
| 1849.71 | 1091.66 | C=O (equatorial) | ||
| 1850.92 | 1950.92 | C=O (axial) | ||
| 1870.38 | 653.08 | C=O | ||
| 1960.72 | 336.00 | C=O | ||
| 3057.72 | 43.54 | CH3 | ||
| 3162.78 | 40.48 | CH3 |
Comparison of Trans and Cis Conformers
The Mo-P bonds are shorter for the trans isomer than in the cis isomer, this is because there is less steric strain in the trans isomer as there is more space for the groups to come closer to the metal. The Mo-C bonds are all the same in the trans isomer but in the cis isomer there are 2 different values depending on whether the C=O groups are round the middle (equatorial) with the phophine groups or above/below (axial) to the phosphine groups. The equatorial C=Os are closer to the metal than in the trans complex and the axial C=Os are further away from the metal centre than in the trans complex. This has the opposite affect of the length of the C-O bonds as the axial C-O bonds are shorter than those in the trans complex and the equatorial C-O bonds are longer than those in the trans complex. This is because bonding to the metal weakens the carbonyl bonding, so the stronger and shorter the M-C bond is, the weaker and longer the C-O bond is. This is due to the degree of metal-carbon backbonding, as more backbonding strengthens the metal-carbon bond but weakens the carbon-oxygen bond. The P-C bonds are a little longer in the cis complex as again due to steric hindrance they can't get as close to each other. The energy differences are very small, only 0.003 a.u. (7.876kJ/mol) between the cis and trans LANL2DZ optimised isomers and 0.009 a.u. (23.629kJ/mol) between the LANL2MB optimised isomers, the cis having the lower energy. There are more C=O peaks in the cis-isomer infrared spectrum then in the trans-isomer. This is because there are different vibrations which will cause changes in dipole as the C=O groups are in different positions compared to the trans-isomer in which the same vibrations would only show one peak as all the C=O groups are in identical positions.
The computer wouldn't let me access the references i wished to use to compare the values obtained from the calculations. The reference i wished to use was Vibrational spectra of the cis and trans isomers of the Mo(CO)4(PPh3)2 complex J. Shamir *, A. Givan, M. Ardon, G. Ashkenazi, Journal of Raman Spectroscopy, Volume 24 Issue 2, Pages 101 - 105
Mini-Project
Fuels of the Futuure
Ammonia Borane is of great interest in the developement of hydrogen fuel. It is a stable molecule with a high hyrdogen content. In the experiment the structure and stability of ammonia borane is to be analysed and compared to similar molcules (ethane) and other methods of releasing hydrogen.
1st Optimisation of Staggered Ammonia Borane

The first optimisation of Ammonia Borane gave a staggered conformation. The B-H bond length was 1.174 angstrom, the N-H bond length was 1.054 angstrom and the B-N bond length was 1.655 angstrom.
The H-B-N bond angle was 104.34° and the H-N-B bond angle was 112.22°.
Summary of optimisation:
File Type: .log
Calculation Type: FOPT
Calculation Method: RB3LYP
Basis Set: LANL2MB
Overall Energy: -82.177 a.u.
Dipole Moment: 5.897 Debye
Point Group: C1
Time Taken for Calculation: 2 mins 38 seconds
2nd Optimsation of Staggered Ammonia Borane
The resulting molecule from the previous optimistion was optimised further by using a better basis set and pseudo-potential. The B-N bond length was 1.685 angstrom, the B-H bond length was 1.212 angstrom and the N-H bond length was 1.022 angstrom. The H-N-B bond angle was 108.75° and the H-B-N bond angle was 105.50°.
Summary of Optimisation
File Type: .log
Calculation Type: FOPT
Calculation Method: RB3LYP
Basis Set: LANL2DZ
Overall Energy: -83.206 a.u.
Dipole Moment: 5.697 Debye
Point Group: C1
Time Taken for Calculation: 5 mins 51.2 seconds
1st Optimisation of Eclipsed Ammonia Borane

The B-N bond length was 1.673 angstrom, the N-H bond length was 1.052 angstrom and the B-H bond length was 1.173 angstrom. The H-N-B bond angle was 112.27° and the H-B-N bond angle was 104.44°.
Summary of Optimisation
File Type: .log
Calculation Type: FOPT
Calculation Method: RB3LYP
Basis Set: LANL2MB
Overall Energy: -82.174 a.u.
Dipole Moment: 5.939 Debye
Point Group: C1
Time Taken for Calculation: 7 mins 7 seconds
The LANL2DZ basis set was also used on this optimised molecule but it reurned a molecule in the staggered conformation so the results were discarded. The following optimisation was run instead.
2nd Optimisation of Eclipsed Ammonia Borane
The B-N bond length was 1.685 angstrom, the N-H bond length was 1.014 angstrom and the B-H bond length was 1.203 angstrom. The H-N-B bond angle was 111.19° and the H-B-N bond angle was 104.73°.
Summary of Optimisation
File Type: .log
Calculation Type: FOPT
Calculation Method: RMP2-FC
Basis Set: 6-31G(d,p)
Overall Energy: -82.920 a.u.
Dipole Moment: 5.654 Debye
Point Group: C1
Time Taken for Calculation: 56 seconds
Comparison of Staggered and Eclipsed Ammonia Borane:
The staggered conformation has an energy of -83.802 a.u compared to -82.920 a.u. of the eclipsed conformation. This results in a energy gap of 2315.691kJ/mol This shows that the staggered conformation has a lower energy and therefore is the more stable and favoured conformation.
Optimisation of Staggered Ethane

C-H bond lengths: 1.1006
C-C bond length: 1.55, 1.77
H-C-C bond angle: 110.74
Summary of Optimsation of Ethane
File Type: .log
Calculation Type: FOPT
Calculation Method: RB3LYP
Basis Set: LANL2MB
Overall Energy: -78.886 a.u.
Dipole Moment: 0.00 Debye
Point Group: C1
Time Taken for Calculation: 47 seconds
Optimisation of Eclipsed Ethane

C-H bond length: 1.10007
C-C bond length: 1.56337
H-C-C bond angle: 111.122
Summary of Optimisation
File Type: .log
Calculation Type: FOPT
Calculation Method: RB3LYP
Basis Set: LANL2MB
Overall Energy: -78.882 a.u.
Dipole Moment: 0.00 Debye
Point Group: C1
Time Taken for Calculation: 37 seconds
Ethane was optimised to provide an organic comparison for ammonia borane.
The energy difference between the staggered and eclipsed conformations of ethane is 10.502kJ/mol. This is alot less than the difference between the ammonia borane conformations (2315.691k/mol). The staggered ammonia borane molecule has a lower energy than the staggered ethane molecule with a difference of 8640.52kJ/mol (comparing the molecules optimised with the same method and basis set.
Bonding
-
 Ammonia Borane HOMO
Ammonia Borane HOMO -
 Ammonia Borane LUMO
Ammonia Borane LUMO -
 Ethane HOMO
Ethane HOMO -
 Ethane LUMO
Ethane LUMO




Ethane is a purely covalent molecule with the C-C bond being formed using an electron from each sp3 hybridised carbon orbital. This results in a neautral molecule with no overall dipole moment.
Ammonia Borane is different as the bond between N (lewis acidic) and B (lewis basic) is a dative covalent bond formed by the lone pairs on nitrogen being donated to the empty p-orbital on boron.
To prove the theory on the bonding previously mentioned, the HOMO and LUMO molecular orbitals have been calculated. It is seen that the ethane molecular orbitals are equal on both carbons showing that the electron density is very similar on both carbons and that the contribution to the bonds must be equal between both carbons.
The Ammonia Borane molecular orbitals are very different as for the HOMO, there is a large electron cloud over boron with a more dense electron cloud over nitrogen, this is due to the lone pairs on the nitrogen which are donated to the B-N bond being held closer to the nitrogen. The LUMO shows a large electron cloud over the nitrogen which shows there is a dipole moment as again the nitrogen is holding the electrons closer to itself as it is more electronegative.
Although this forms a weaker bond than the covalent bond in ethane, its is quite highly polarised, which improves intermolecular interactions.
This results in ammonia borane having a much higher melting point than ethane as the nitrogen hydrogens are acidic (as the nitrogen is more electronegative) and the boron hydrogens are hydridic. This allows the molecules to form electrostatic dihydrogen bonds between each other to form a orthorhombic network resulting in a crystal structure.
Comparison with other possible reactants
NaBH4 and NH4Cl:


NaBH4 and NH4Cl were both optimised as BH4- and NH4+. The resulting energies were much higher than that of ammonia borane, showing that ammonia borane is much more stable than the reactants used for another method of hydrogen production. NH4BH4 was attampted to be optimised but it didn't work as the molecule ended up as NH3BH3, this shows that NH4BH4 is very unstable, which means it may release hydrogen with more ease but it isn't safe to store.
Conclusion
Overall it has been seen from the previous calculations that Ammonia Borane is a stable molecule, more stable than it organic comparison, which shows that it is a good method of storing hydrogen. It may not be the most efficient molecule for releasing hydrogen but other molecules that may release hydrogen easier are generally less stable. Further things to be looked into are other possibilities of releasing hydrogen for comparison with ammonia borane and how easily ammonia borane can release hydrogen. From the amount of research done in this project, it has been concluded that ammonia borane is a good reactant for hydrogen fuel.
References
1. Spectroscopic studies of the phase transition in ammonia borane: Raman spectroscopy of single crystal NH[sub 3]BH[sub 3] as a function of temperature from 88 to 330 K
Nancy J. Hess, Mark E. Bowden, Vencislav M. Parvanov, Chris Mundy, Shawn M. Kathmann, Gregory K. Schenter, and Tom Autrey, J. Chem. Phys. 128, 034508 (2008), DOI:10.1063/1.2820768
2. Room-Temperature Structure of Ammonia Borane
Mark E. Bowden, Graeme J. Gainsford, and Ward T. Robinson, Aust. J. Chem. 2007, 60, 149–153
3. Bowden, M., T. Autrey, et al. (2008). "The thermal decomposition of ammonia borane: A potential hydrogen storage material." Current Applied Physics 8(3-4): 498-500.
4.Synthesis of ammonia borane for hydrogen storage applications. David J. Heldebrant, Abhi Karkamkar, John C. Linehan and Tom Autrey, Energy Environ. Sci., 2008, 1, 156 - 160, DOI:0.1039/b808865a 5.