
Rep:Mod:rebeccalee
Introduction
Computational chemistry is an important way to analyze molecules that either cannot be characterized in the wet lab, or as a safer way to find properties. It is able to optimize molecules, thus giving information such as geometry, and also give other information such as IR spectra.
For this experiment, GaussView 5.0 is used to optimize and analyze various molecules by using a variety of methods and basis sets. HPC was used when a more powerful computational method was needed.
In an optimization calculation, two things occur: 1. The nuclei positions of each atom (B and H's) are assumed to be fixed in order to solve the Schrodinger equation for the electrons. This is known as the SCF part of the optimization. 2. The nuclei position is changed and the SCF is repeated as the geometry changes until the geometry with the lowest energy is obtained. This is the OPT part of the optimization.
The energy values obtained typically have an error of about 10 kJ/mol. Since 1 a.u. = 2625.50184 kJ/mol, this error is about 0.004 a.u., and thus energies are reported to 0.01 a.u.. All dipole moments are reported to 0.01 Debye, frequencies and intensities to the nearest whole integer, bond distances to about 0.01Å, and bond angles to about 0.1°.
For all vibrational analysis data, the link above each photo will give a link to the animation of the described movement of the atoms.
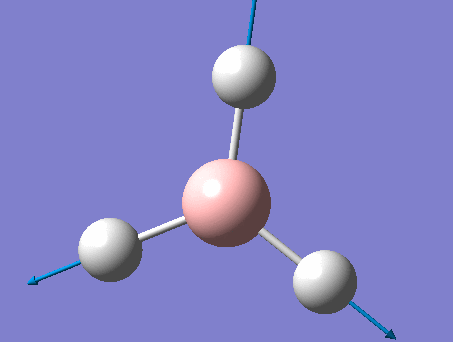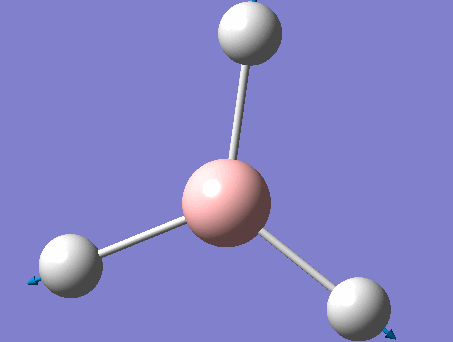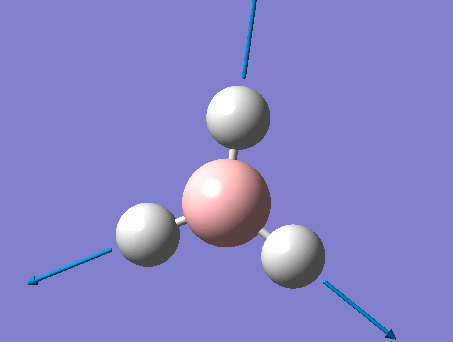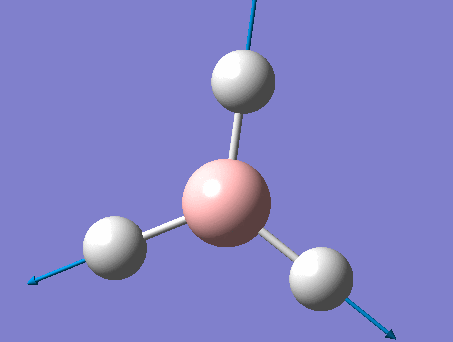
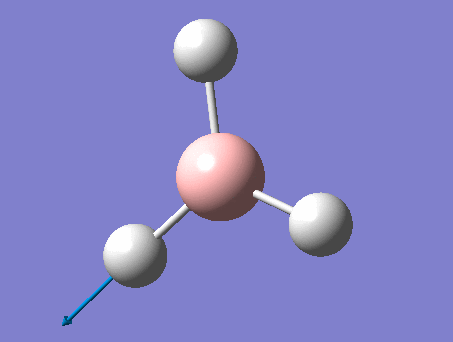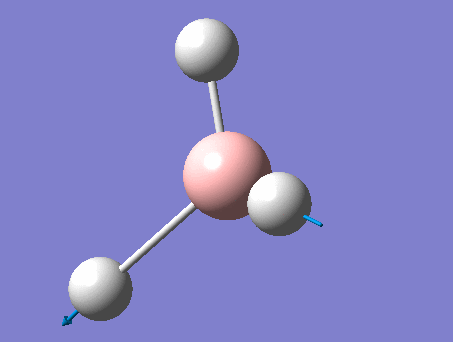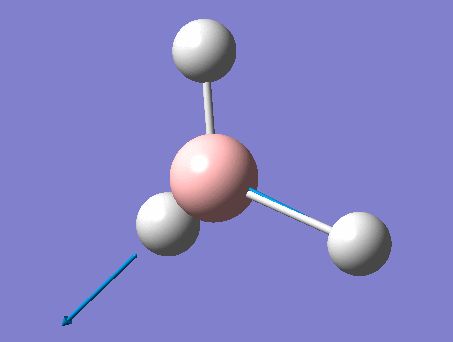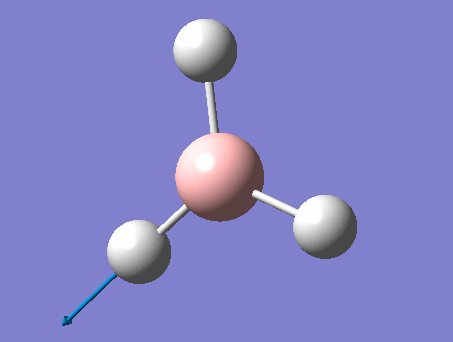
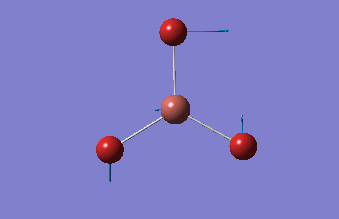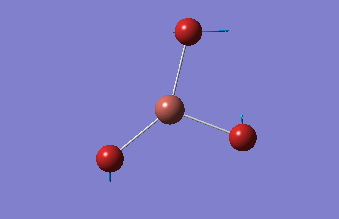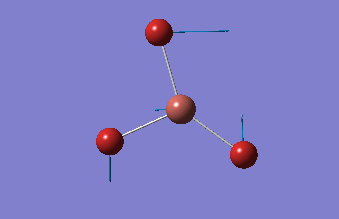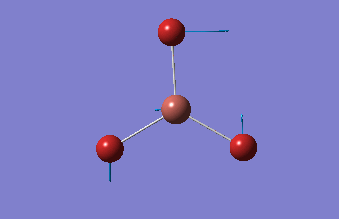
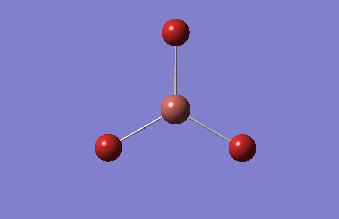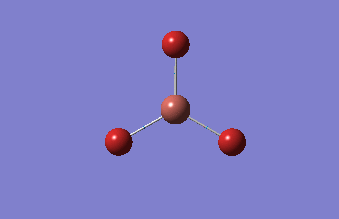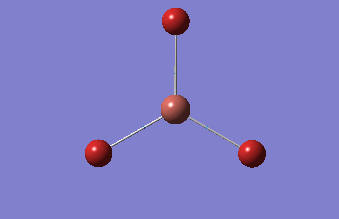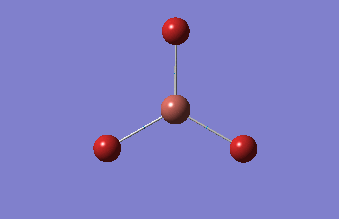
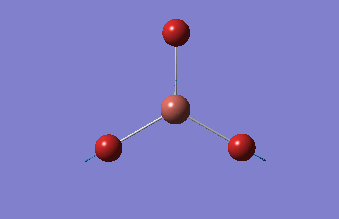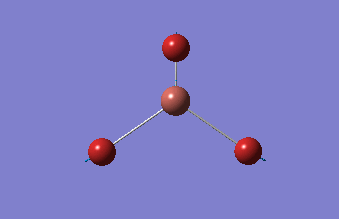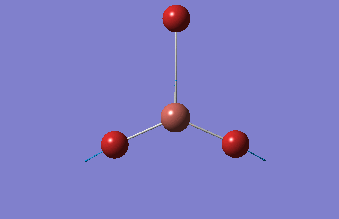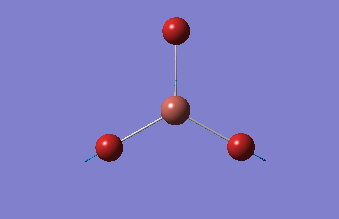
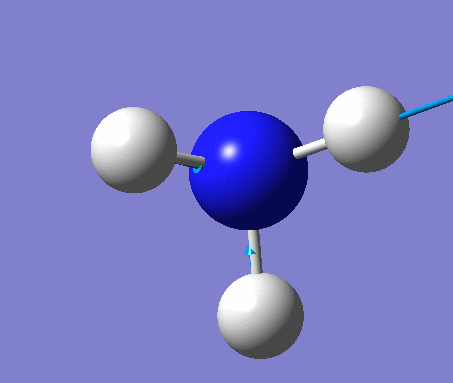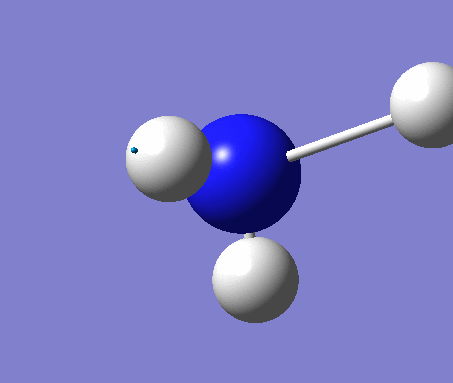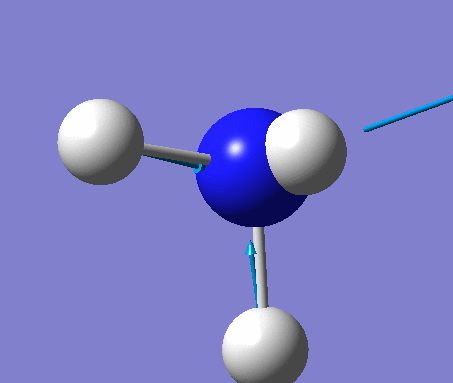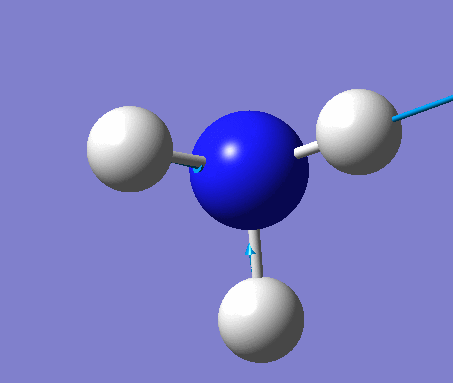
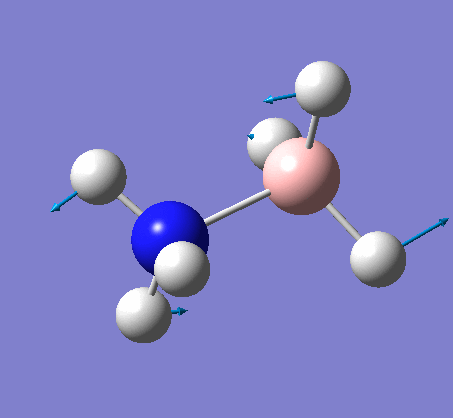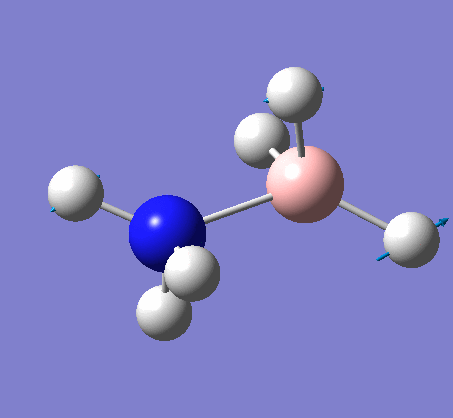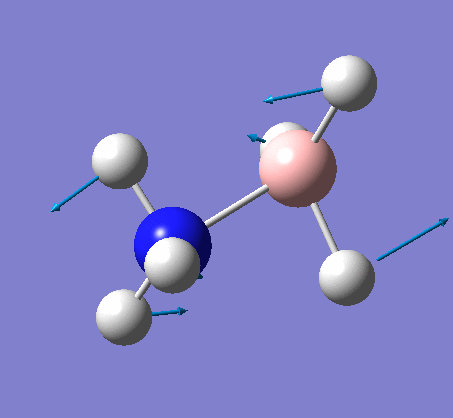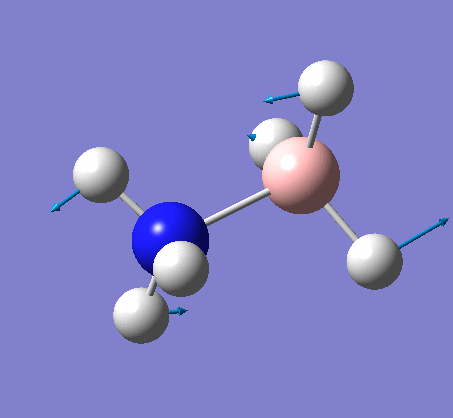
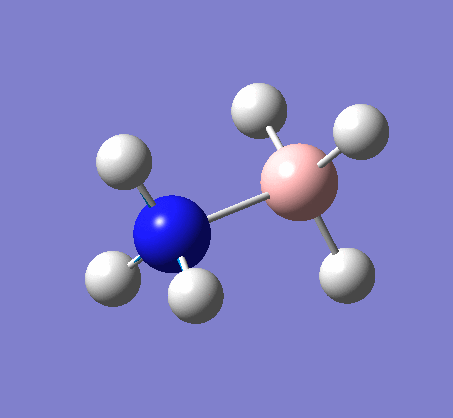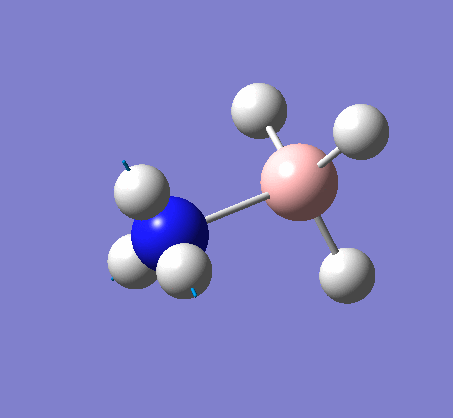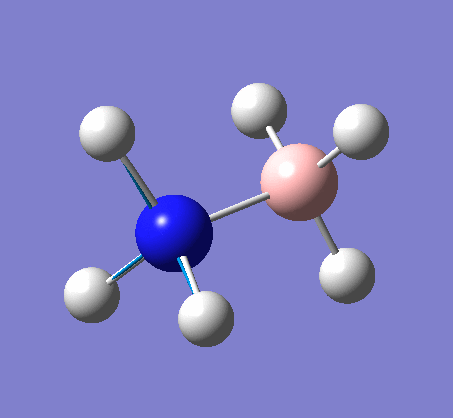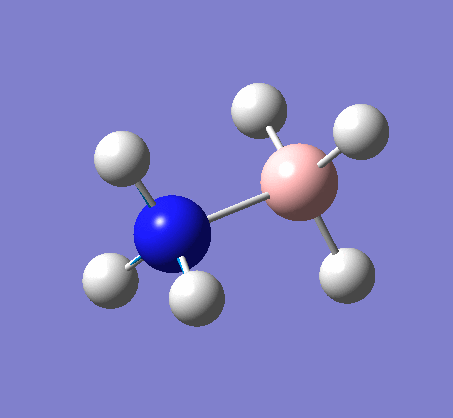
BH3
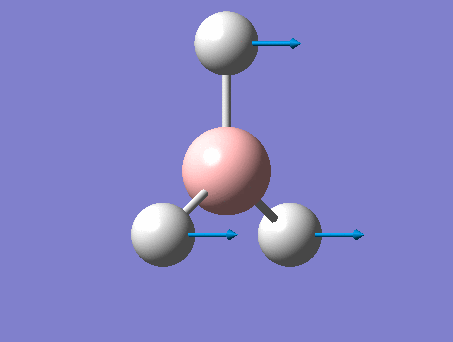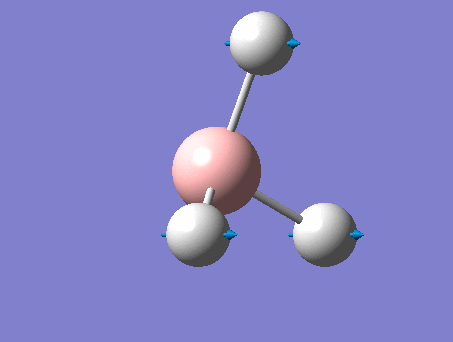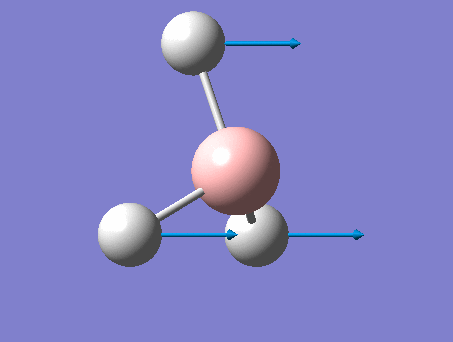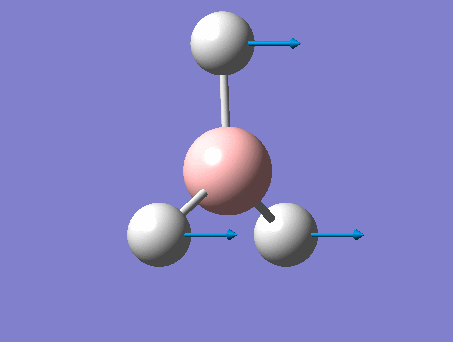
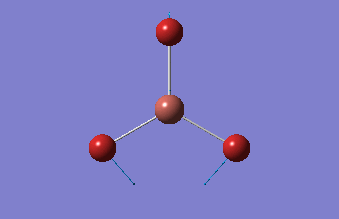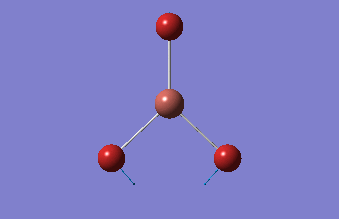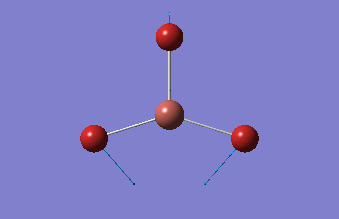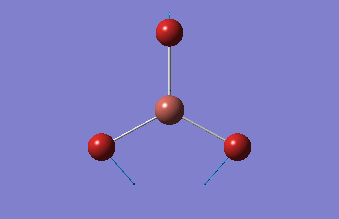
BH3 was drawn with trigonal planar geometry, B-H bond lengths of 1.50Å, and H-B-H bond angles of 120.0°.
3-12G optimization
Optimization was achieved using the DFT/B3LYP method, 3-12G basis set and optimization job. Because the 3-12G basis set has very low accuracy, the time taken for this calculation was very short. Since BH3 is a symmetrical and simple molecule, the use of this basis set is justified.
This proceeded to give the following .log file: File:Rl bh3opt.log.

| B-H length | H-B-H angle |
|---|---|
 |

|
| 1.19Å | 120.0° |
In the .log file, there is a section that shows whether or not the parameters converged. It is always important to check this after each calculation to ensure that the parameters did converge. Here, the optimization was successful, and the following data from the log file shows that all the parameters did not fail to converge, and also shows the optimized bond lengths and angles:
Item Value Threshold Converged?
Maximum Force 0.000413 0.000450 YES
RMS Force 0.000271 0.000300 YES
Maximum Displacement 0.001610 0.001800 YES
RMS Displacement 0.001054 0.001200 YES
Predicted change in Energy=-1.071764D-06
Optimization completed.
-- Stationary point found.
----------------------------
! Optimized Parameters !
! (Angstroms and Degrees) !
-------------------------- --------------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------------------
! R1 R(1,2) 1.1935 -DE/DX = 0.0004 !
! R2 R(1,3) 1.1935 -DE/DX = 0.0004 !
! R3 R(1,4) 1.1935 -DE/DX = 0.0004 !
! A1 A(2,1,3) 120.0 -DE/DX = 0.0 !
! A2 A(2,1,4) 120.0 -DE/DX = 0.0 !
! A3 A(3,1,4) 120.0 -DE/DX = 0.0 !
! D1 D(2,1,4,3) 180.0 -DE/DX = 0.0 !
--------------------------------------------------------------------------------
GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
The following two graphs show the optimization against energy and gradient:
| Total energy graph | Gradient graph |
|---|---|
 |

|
These graphs also indicate that after four iterations the optimization was completed. The total energy graph shows gaussview finding the structure of BH3 in comparison to the potential energy. The energy decreases as the B-H bond length gets closer to the equilibrium bond length value. The root mean squared gradient is very close to zero, which indicates a successful optimization. It indicates that the structure found is at a minimum.
The data above shows that the optimized BH3 molecule has B-H bond lengths of 1.19Å, and H-B-H bond angles of 120.0°. This is in agreement with literature.[1] The symmetry point group is D3h, and has a 0.00 Debye dipole moment due to its high symmetry. The final energy is -26.46226338 a.u., and took 8.0 seconds to run.
6-31G optimization
This time, optimization was achieved using the DFT/B3LYP method and 6-31G(d,p) basis set. This is a higher quality basis set which means there is better accuracy with this optimization. However, because the basis set and method is different to the initial optimization, this means that these two results cannot be compared.
This gave the .log file: File:Rl bh3opt631g.log

| B-H length | H-B-H angle |
|---|---|
 |

|
| 1.19Å | 120.0° |
The optimization was successful, and the following data shows that all the parameters converged:
Item Value Threshold Converged?
Maximum Force 0.000433 0.000450 YES
RMS Force 0.000284 0.000300 YES
Maximum Displacement 0.001702 0.001800 YES
RMS Displacement 0.001114 0.001200 YES
Predicted change in Energy=-1.189019D-06
Optimization completed.
-- Stationary point found.
----------------------------
! Optimized Parameters !
! (Angstroms and Degrees) !
-------------------------- --------------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------------------
! R1 R(1,2) 1.1914 -DE/DX = 0.0004 !
! R2 R(1,3) 1.1914 -DE/DX = 0.0004 !
! R3 R(1,4) 1.1914 -DE/DX = 0.0004 !
! A1 A(2,1,3) 120.0 -DE/DX = 0.0 !
! A2 A(2,1,4) 120.0 -DE/DX = 0.0 !
! A3 A(3,1,4) 120.0 -DE/DX = 0.0 !
! D1 D(2,1,4,3) 180.0 -DE/DX = 0.0 !
--------------------------------------------------------------------------------
GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
The following two graphs show the optimization against energy and gradient:
| Total energy graph | Gradient graph |
|---|---|
 |

|
These graphs also indicate that after four iterations the optimization was completed. The total energy graph shows gaussview finding the structure of BH3 in comparison to the potential energy. The energy decreases as the B-H bond length gets closer to the equilibrium bond length value. The root mean squared gradient is very close to zero, which indicates a successful optimization. It indicates that the structure found is at a minimum.
The data above shows that the optimized BH3 molecule has B-H bond lengths of 1.19Å, and H-B-H bond angles of 120.0°. The symmetry point group is still D3h, and again has a 0.00 Debye dipole moment due to its high symmetry. The resultant energy is -26.61532252 a.u., which is close to the energy from the previous optimization, and took 10.0 seconds to run.
| 3-21G | 6-31G |
|---|---|
| -26.46226338 a.u. | -26.61532252 a.u. |
The gives an energy difference of 0.1530592 a.u., which is 401.86 kJ/mol.
Even though in both these optimizations the stationary point has been found, it does not prove that the parameters converged to a minimum. In order to ensure that this has happened, the frequency and vibrational analysis must be run.
If at a minima, then any relative movement of atoms will give a higher energy state, which means energy must be given. If at a maxima, this will give the opposite effect in that energy will be released when this happens. Thus this data can be used to distinguish between a minima or maxima convergence. All the vibrational frequencies for a minima will be positive, whereas there will be one negative frequency if at a maxima. If in the case of more than one negative frequency, this would then mean that a stationary point was not actually found.
Frequency analysis
This analysis, along with the vibrational analysis, can give a prediction of the IR spectrum which can then be compared to the data from the wet lab. The second use is as explained before. The second derivative of the graph will be checked in order to see if the minima was indeed found. Using the same method as for the optimization, but changing the job type to frequency.
The same method was used, just changing the job to frequency.
This frequency analysis gave the following .log file: File:Rl bh3freq.log. The data below shows that the minima was found.

| B-H length | H-B-H angle |
|---|---|
 |

|
| 1.19Å | 120.0° |
The frequency calculation was successful, and the following data shows that all the parameters converged:
Item Value Threshold Converged?
Maximum Force 0.000433 0.000450 YES
RMS Force 0.000217 0.000300 YES
Maximum Displacement 0.001697 0.001800 YES
RMS Displacement 0.000848 0.001200 YES
Predicted change in Energy=-1.103020D-06
Optimization completed.
-- Stationary point found.
GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
The data below shows that the minima was found and that the results are quite accurate as the low frequencies are close to 0. The first line of low frequencies corresponds to the "-6" vibrational modes (every molecule has 3N-6 vibrational modes). The closer these values are to 0, the better the accuracy of the results. These values should always be smaller than the ones in the second row. The first row of the low frequencies should ideally be within 10 cm-1 of each other. However, this is not the case here showing that the basis set used is not accurate enough. Thus next time a more accurate basis set should be used. Since the second row of low frequencies does not give any negative values, this means that the optimization was successful and gave a minimum.
Low frequencies --- -70.3431 -69.5542 -69.5540 -0.0055 0.0695 0.1595 Low frequencies --- 1161.4019 1212.1084 1212.1111
Vibrational analysis
| No. | Vibration form | Frequency (cm-1) | Literature value of frequency (cm-1)[1] | Intensity | Symmetry (D3h point group) | Description |
|---|---|---|---|---|---|---|
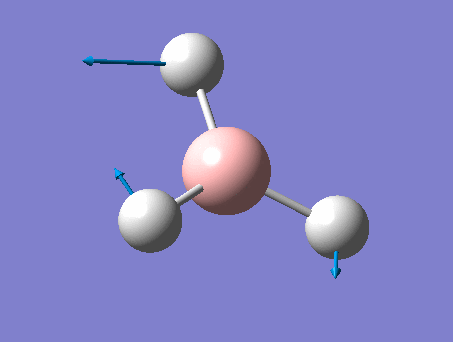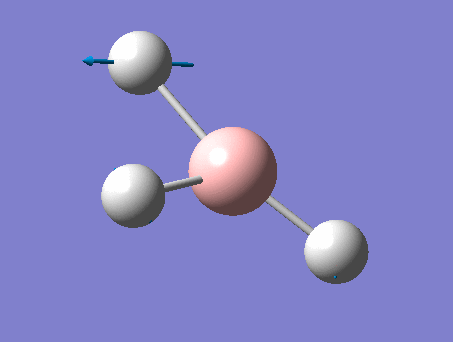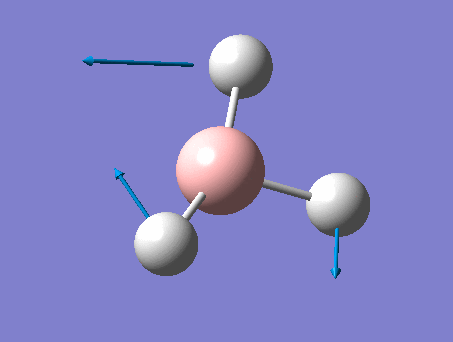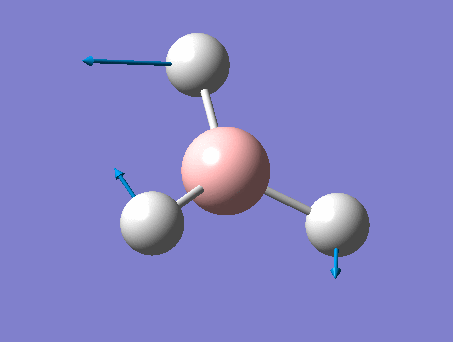
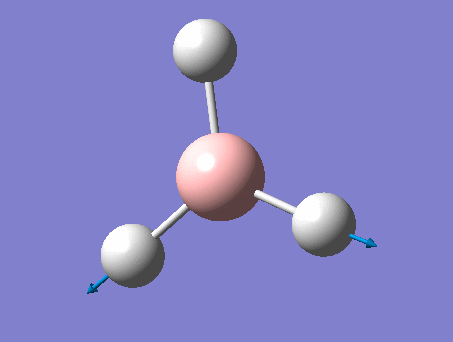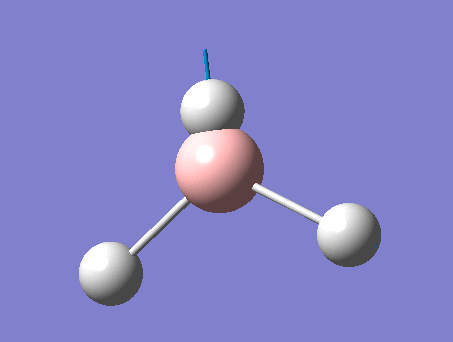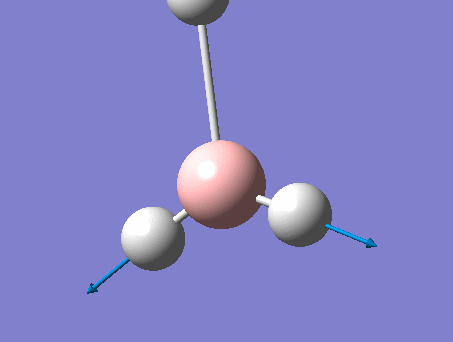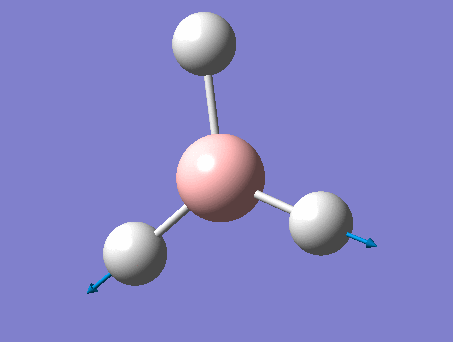
| 1 | Wagging  |
1161 | 1148 | 93 | A2" | All three H's move in one direction in a concerted motion. The B moves in the opposite direction of these H's at all times. Due to this, there is a large change in dipole moment causing an intense peak. This is also known as the umbrella wagging motion. |
| 2 | Scissoring  |
1212 | 1197 | 14 | E' | Two of the H's move down at the same time, while the other H and B move up slightly to counteract this motion. This means that one of the H-B-H angle's is contracted while the other two are expanded. |
| 3 | In plane rocking  |
1212 | 1197 | 14 | E' | Two of the H's move as in the scissoring motion whilst the other moves a lot in the opposite direction in order to oppose this motion. Thus one H-B-H angle is contracted, one expanded, and the third held constant. |
| 4 | Symmetric stretching  |
2588 | not observed | 0 | A1' | A symmetric B-H stretching of all three at the same time. The central B does not move during this movement. Thus there is on change in dipole moment and this peak is not observed. |
| 5 | Asymmetric stretching  |
2722 | 2808 | 126 | E' | One B-H length stays constant whilst the other two lengthen and shorten at the same time. |
| 6 | Asymmetric stretching  |
2722 | 2808 | 126 | E' | Two of the B-H bonds stretch symmetrically, whilst the third one stretches asymmetrically relative to the other two. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
This gives the corresponding IR spectrum:

Modes 2 and three are degenerate, which gives only one peak in the IR spectrum. This is the same for modes 5 and 6. Mode 4 has an infrared value of 0, thus it does not give a peak in the spectrum. The reason why it has this value is because its overall dipole moment does not change as the stretch is completely symmetric. This is why although there are 6 vibrational modes for BH3, there are only 3 peaks in the spectrum.
BH3 molecular orbitals
Another calculation with the same method was run, except this time changing the job to energy. The .chk file was used to do MO analyis. The link to D-space is: [[2]]. The following steps were followed while forming this diagram [2]:
1. Determine the shape, point group, and define the axial system.
2. Find all the symmetry elements of this molecule.
3. Identify and put the chemical fragments on the bottom of the diagram.
4. Determine the energy levels and symmetry labels of the fragment molecules.
5. Combine fragment molecules of the same symmetry, estimate the character and extent of splitting, and draw the molecular orbitals.
6. Determine the number of electrons of each fragment and add this to the diagram, and determine if any mixing occurs.

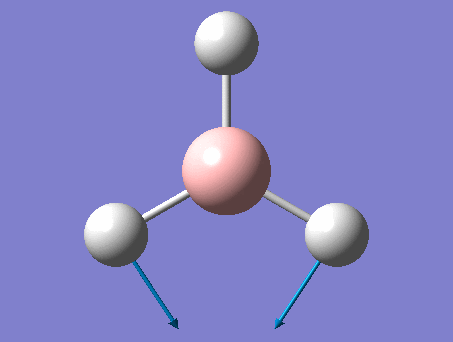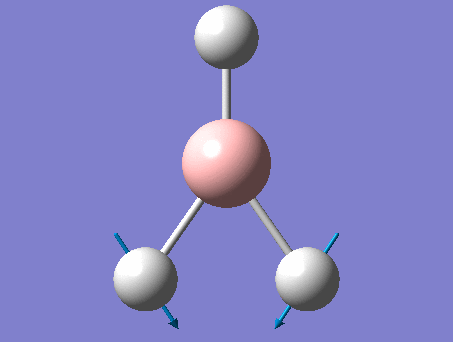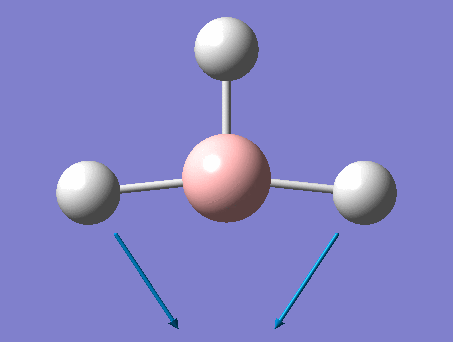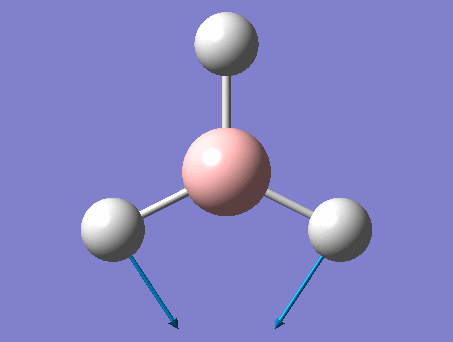
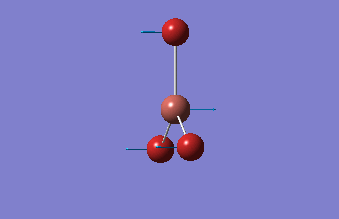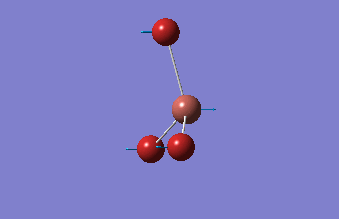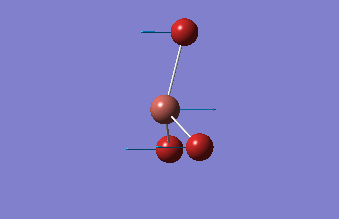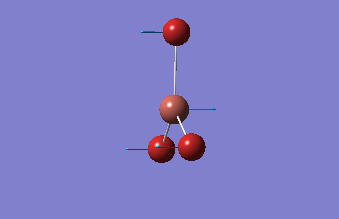
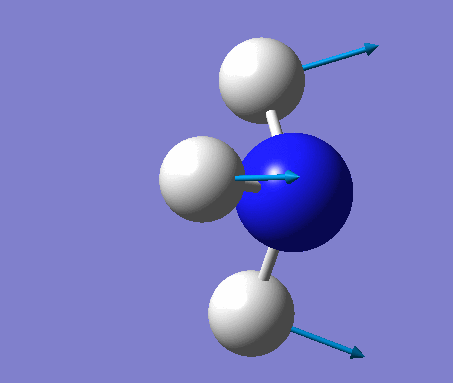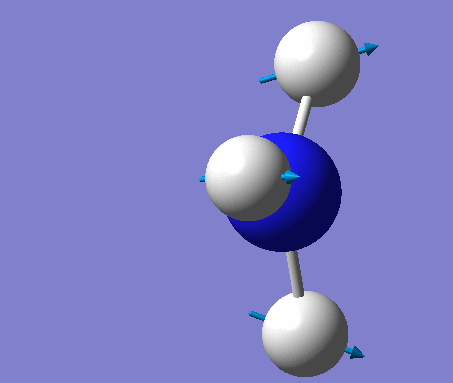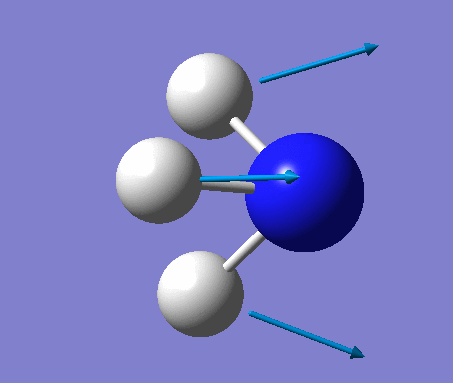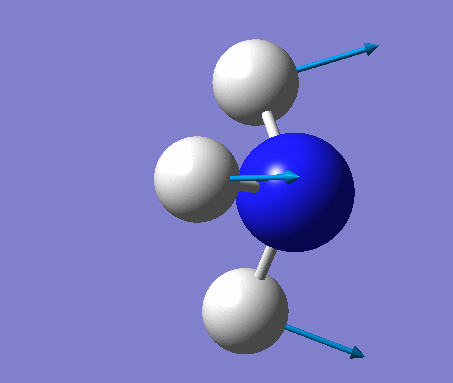
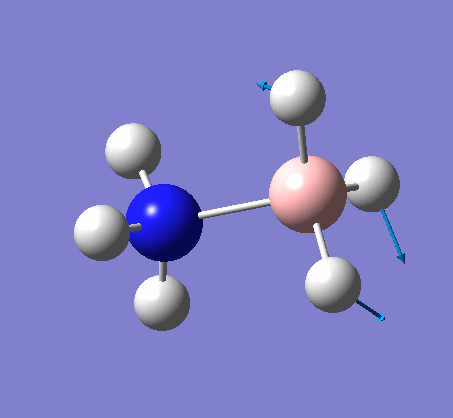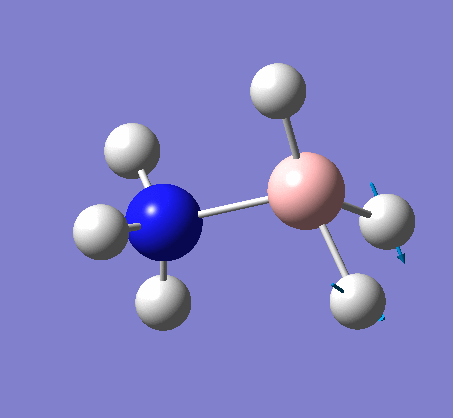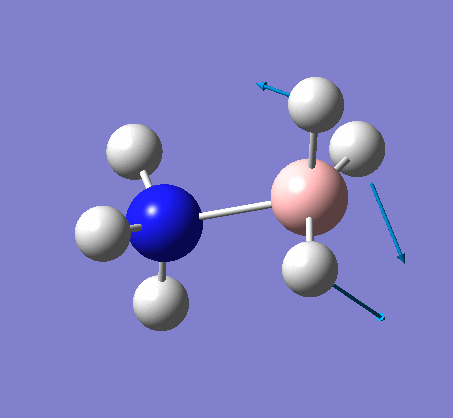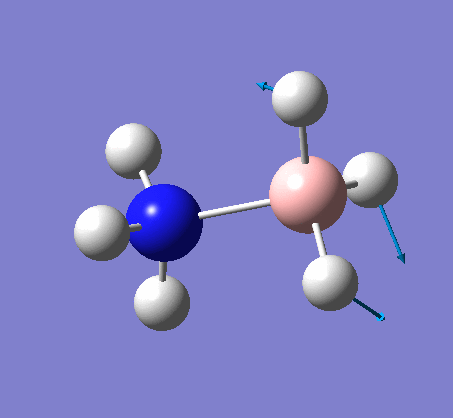
The shape of this molecule is trigonal planar, with a D3h point group. The highest rotation axis is chosen as the z axis, thus the molecule lies in the xy plane. See the figure to the right for the symmetry elements and axial system. The two fragments are the B atom and H3.
The totally bonding orbital of H3 is symmetric and thus has a'1 symmetry. The degenerate orbitals are e' according to the character table. The boron s orbitals are always symmetric and thus a'1 symmetry. The rest of the orbitals are compared with the x, y, and z axis and characterized according to the character table. The energy levels of the s orbitals of the B and H3 fragments are similar due to similar electronegativities, but the all bonding H3 fragment is slightly stabilized and thus has a lower energy. After all these are drawn out and labeled, LCAO is used and the resultant orbitals are drawn.
This gives the following diagram:

It is difficult to decide if the 2e' level is higher than the 3a'1 level, however the gaussian calculation puts it in the order seen. Here it seems the s-s interactions are weaker than the s-p thus the 3a'1 level is lower than the 2e'.
There do not seem to be great differences between LCAO and real MOs, which shows that qualitative MO theory is accurate and very useful. This means gaussian can be used to calculate these MOs in a reliable manner, as long as the correct methods and basis sets are used.
TlBr3
TlBr3 was first drawn with trigonal planar geometry, and constrained to the D3h point group. This analysis shows the importance of computational chemistry as Tl is highly toxic and thus its properties can be investigated without risk.
LANL2DZ optimization
Optimization was achieved using the DFT/B3LYP method and LANL2DZ basis set. The LANL2DZ is a medium level basis set and thus has a bit more accuracy than the previous two basis sets used.
The completed optimization was submitted to D-space: [3]

| Tl-Br length | Br-Tl-Br angle |
|---|---|
 |

|
| 2.65Å | 120.0° |
The optimization was successful, and the following data shows that all the parameters converged:
Item Value Threshold Converged?
Maximum Force 0.000002 0.000450 YES
RMS Force 0.000001 0.000300 YES
Maximum Displacement 0.000022 0.001800 YES
RMS Displacement 0.000014 0.001200 YES
Predicted change in Energy=-6.084106D-11
Optimization completed.
-- Stationary point found.
----------------------------
! Optimized Parameters !
! (Angstroms and Degrees) !
-------------------------- --------------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------------------
! R1 R(1,2) 2.651 -DE/DX = 0.0 !
! R2 R(1,3) 2.651 -DE/DX = 0.0 !
! R3 R(1,4) 2.651 -DE/DX = 0.0 !
! A1 A(2,1,3) 120.0 -DE/DX = 0.0 !
! A2 A(2,1,4) 120.0 -DE/DX = 0.0 !
! A3 A(3,1,4) 120.0 -DE/DX = 0.0 !
! D1 D(2,1,4,3) 180.0 -DE/DX = 0.0 !
--------------------------------------------------------------------------------
GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
The following two graphs show the optimization against energy and gradient:
| Total energy graph | Gradient graph |
|---|---|
 |

|
These graphs indicate that after three iterations the optimization was completed.
The optimized bond length is 2.65Å, and bond angle is 120.0°. This is close to the literature value of 2.51Å. [3] The resultant energy is -91.21812851 a.u., and took 37.4 seconds to run. Again there is a 0.00 Debye dipole moment as the molecule is symmetric.
Frequency analysis
This frequency analysis was submitted to D-space: [4]. The same method as for the optimization was run, but the job was changed to frequency.

| Tl-Br length | Br-Tl-Br angle |
|---|---|
 |

|
| 2.65Å | 120.0° |
The frequency calculation was successful, and the following data shows that all the parameters converged and that the minimum was found:
Item Value Threshold Converged?
Maximum Force 0.000002 0.000450 YES
RMS Force 0.000001 0.000300 YES
Maximum Displacement 0.000022 0.001800 YES
RMS Displacement 0.000011 0.001200 YES
Predicted change in Energy=-5.660901D-11
Optimization completed.
-- Stationary point found.
GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
Low frequencies --- -3.4213 -0.0026 -0.0004 0.0015 3.9367 3.9367 Low frequencies --- 46.4289 46.4292 52.1449
Vibrational analysis
| No. | Vibration form | Frequency (cm-1) | Literature value of frequency (cm-1)[4] | Intensity | Symmetry (D3h point group) | Description |
|---|---|---|---|---|---|---|
| 1 | Wagging  |
46 | 47 | 4 | E' | All three H's move in one direction in a concerted motion. The B moves in the opposite direction of these H's at all times. Due to this, there is a large change in dipole moment causing an intense peak. This is also known as the umbrella wagging motion. |
| 2 | Scissoring  |
46 | 47 | 4 | E' | Two of the H's move down at the same time, while the other H and B move up slightly to counteract this motion. This means that one of the H-B-H angle's is contracted while the other two are expanded. |
| 3 | In plane rocking  |
52 | 63 | 6 | A2" | Two of the H's move as in the scissoring motion whilst the other moves a lot in the opposite direction in order to oppose this motion. Thus one H-B-H angle is contracted, one expanded, and the third held constant. |
| 4 | Symmetric stretching  |
165 | 185 | 0 | A1' | A symmetric B-H stretching of all three at the same time. The central B does not move during this movement. Thus there is on change in dipole moment and this peak is not observed. |
| 5 | Asymmetric stretching  |
211 | 203 | 25 | E' | One B-H length stays constant whilst the other two lengthen and shorten at the same time. |
| 6 | Asymmetric stretching  |
211 | 203 | 25 | E' | Two of the B-H bonds stretch symmetrically, whilst the third one stretches asymmetrically relative to the other two. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
- Note the literature values are of TlBr3-LiBr. Thus the values are just used for a comparison.
This gives the corresponding IR spectrum:

Modes 2 and three are degenerate, which gives only one peak in the IR spectrum. This is the same for modes 5 and 6. Mode 4 has an infrared value of 0, thus it does not give a peak in the spectrum. The reason why it has this value is because its overall dipole moment does not change as the stretch is completely symmetric. This is why although there are 6 vibrational modes for TlBr3, there are only 3 peaks in the spectrum.
The lowest 'real' mode is 46 cm-1.
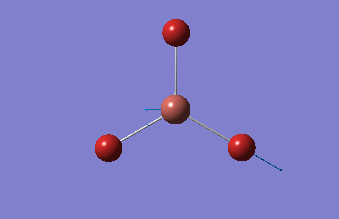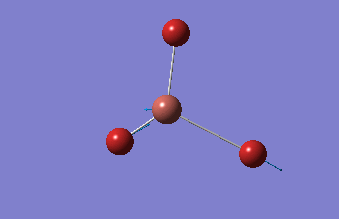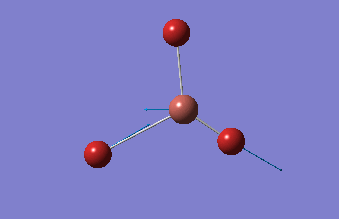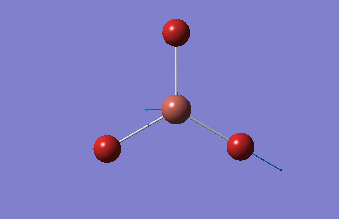
BBr3
BBr3 was first drawn with trigonal planar geometry, and constrained to the D3h point group.
Optimization
Optimization was achieved using the DFT/B3LYP method and GEN basis set.
The completed optimization was submitted to D-space: [5]

| B-Br length | Br-B-Br angle |
|---|---|
 |

|
| 1.93Å | 120.0° |
The optimization was successful, and the following data shows that all the parameters converged:
Item Value Threshold Converged?
Maximum Force 0.000008 0.000450 YES
RMS Force 0.000005 0.000300 YES
Maximum Displacement 0.000036 0.001800 YES
RMS Displacement 0.000023 0.001200 YES
Predicted change in Energy=-4.026780D-10
Optimization completed.
-- Stationary point found.
----------------------------
! Optimized Parameters !
! (Angstroms and Degrees) !
-------------------------- --------------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------------------
! R1 R(1,2) 1.934 -DE/DX = 0.0 !
! R2 R(1,3) 1.934 -DE/DX = 0.0 !
! R3 R(1,4) 1.934 -DE/DX = 0.0 !
! A1 A(2,1,3) 120.0 -DE/DX = 0.0 !
! A2 A(2,1,4) 120.0 -DE/DX = 0.0 !
! A3 A(3,1,4) 120.0 -DE/DX = 0.0 !
! D1 D(2,1,4,3) 180.0 -DE/DX = 0.0 !
--------------------------------------------------------------------------------
GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
The following two graphs show the optimization against energy and gradient:
| Total energy graph | Gradient graph |
|---|---|
 |

|
These graphs indicate that it took three iterations to complete the job.
The optimized bond length is 1.93Å, and bond angle of 120.0°. This bond length is in close agreement with the literature. [5] The dipole moment is still 0.00 Debye as the molecule is symmetric, and the resultant energy is -64.43645296 a.u.. The method took 35.8 seconds to run.
Structural comparisons
| molecule | length (Å) |
|---|---|
| BH3 | 1.19 |
| TlBr3 | 2.65 |
| BBr3 | 1.93 |
Boron and thallium are both in group 3, however B is in period 2 and Tl in period 6. This means Tl has more diffuse valence orbitals, and thus poorer overlap with bromine than B does. Thus Tl-Br bond lengths will be greater than B-Br bonds.
Br is less electronegative than H, and it is also a much larger ligand. Thus it would be expected that the B-Br bond lengths would be longer than those for B-H due to poorer overlap. Thus changing the ligand changes the properties of the bond, including the bond length.
Br and H are both non-metals but with different electronegativities. The electronegativity difference between Br/B and H/B is higher for the former. Thus B-H bonds are less polar than the B-Br bonds.
Gaussview determines whether or not a bond exists based upon the distance between the two atoms. Generally this approximation works well, however if gaussview decides the distance is too long for a bond to exist then it will not show the bond. However it is quite often shown that there is significant bonding interactions even at these lengths, and thus this is a shortcoming of gaussview. In reality a bond is an attractive force that keeps two atoms/substituents together. In this bond, the electron density is very high, thus measurement of electron density rather than bond lengths could be a better approximation for gaussview.
Comparison of BH3 and TlBr3 vibrational analyses
| mode | BH3 frequency (cm-1) | TlBr3 frequency (cm-1) |
|---|---|---|
| 1 | 1161 | 46 |
| 2 | 1212 | 46 |
| 3 | 1212 | 52 |
| 4 | 2589 | 165 |
| 5 | 2722 | 211 |
| 6 | 2722 | 211 |
Hooke's law gives an approximation of an absorption frequency: ν = (1/c2pi) * (f/μ)^(1/2), where μ is the reduced mass, f is the force constant of the bond, c is the speed of light. Thus, this gives a clear explanation as to why TlBr3 frequencies are a lot smaller than those for BH3. The masses of Tl and Br are much more than those of B and H, thus according to this equation the frequency is inversely proportional to the reduced mass. Because TlBr3 is so much heavier, its corresponding frequencies are a lot smaller.
These two spectra are similar in the sense that there are 6 vibrational modes, but only 3 peaks in the spectra due to 2 sets of degenerate modes, 1 active mode, and 1 inactive mode. The symmetry and geometry of the molecules are the same so the difference is not extreme. For both of the spectra, the A2" and E' modes are close together in energy, A1' and E modes are close together. However, in BH3 the A2" mode is lower than the E' modes, whereas the order is the other way around for TlBr3. This reordering occured since the TlBr3 A2" vibration has a greater dipole moment than that for BH3. The dipole moment is larger due to the more polarizable electron cloud of TlBr3, thus the distortion is greater when the molecule oscillates.
The same method and basis set must be used for both optimization and frequency analysis calculations in order to ensure that the results can be compared. If anything is changed then the calculated molecule will give different values and thus the accuracy of the analysis cannot be ensured. The frequency analysis is run in order to ensure that the optimized molecule is at the minimum. The low frequencies help indicate if the frequency job was run correctly, i.e. no negative frequencies, and indicate the accuracy of the said calculation. This is in the first line of the low frequencies. The closer to 0 these frequencies are and the closer the values are to each other, the better the accuracy. If it is not this way, then a new basis set must be used to increase this accuracy.
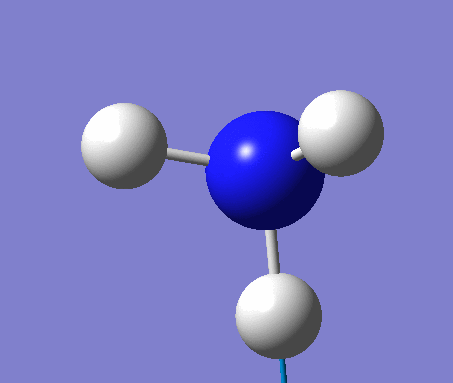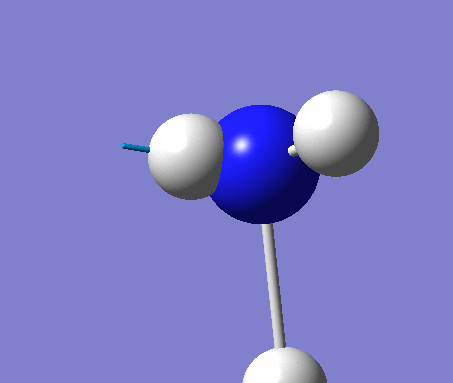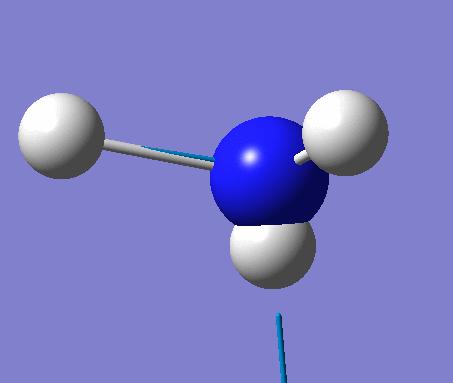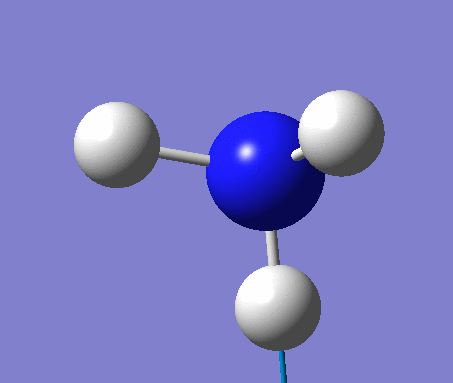
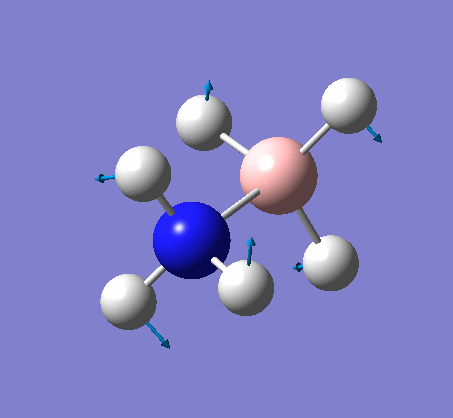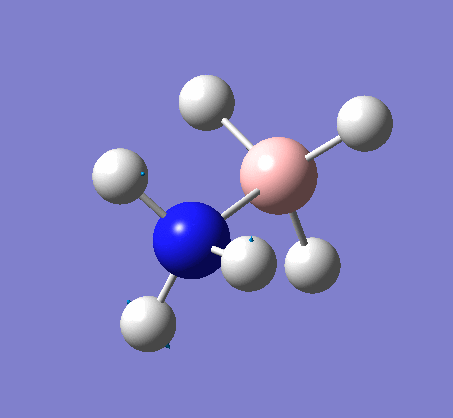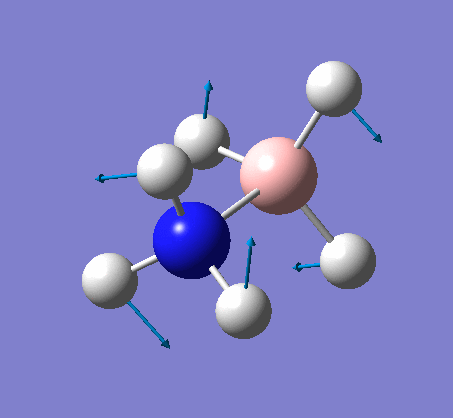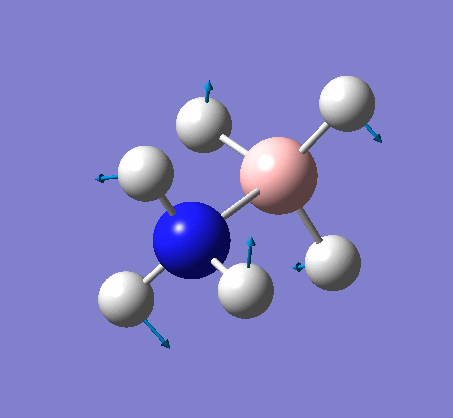
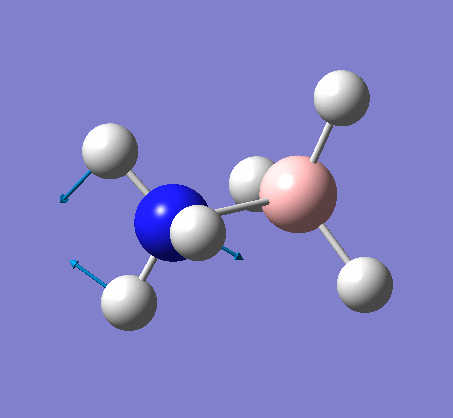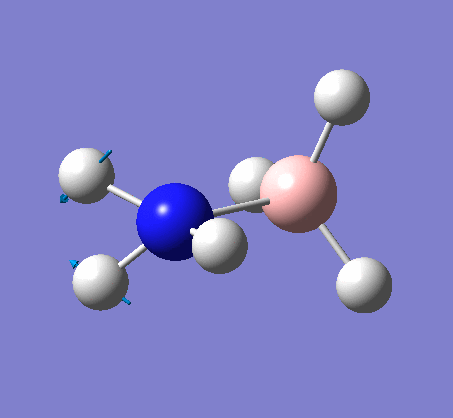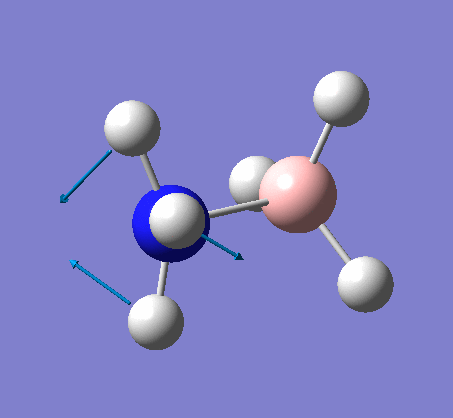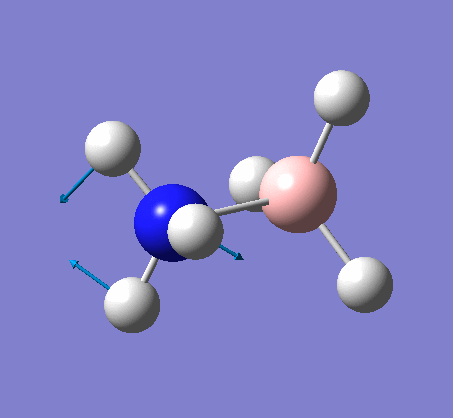
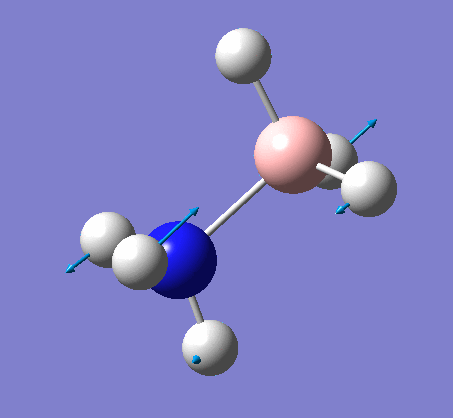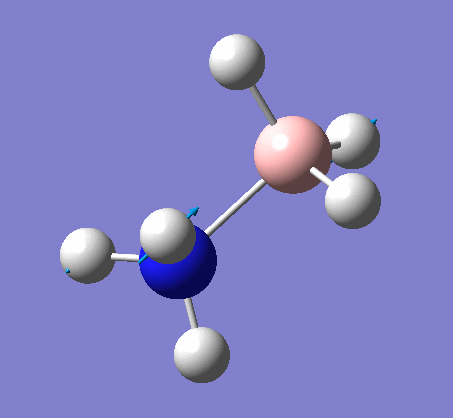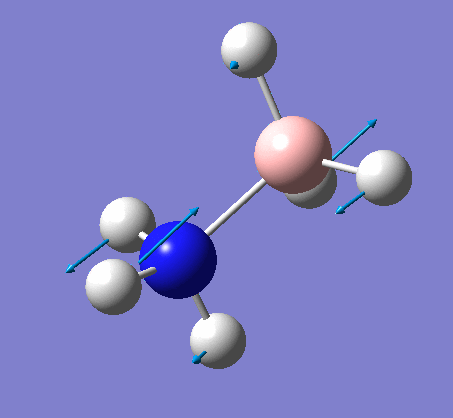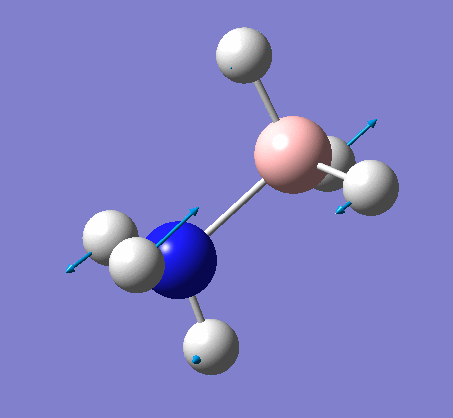
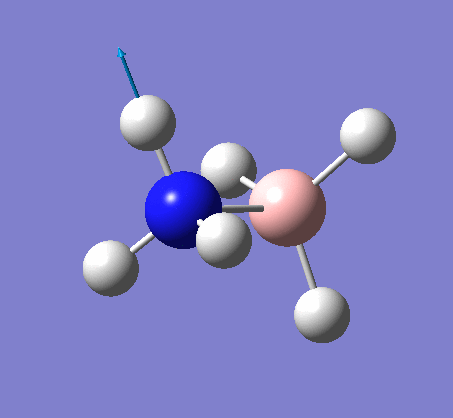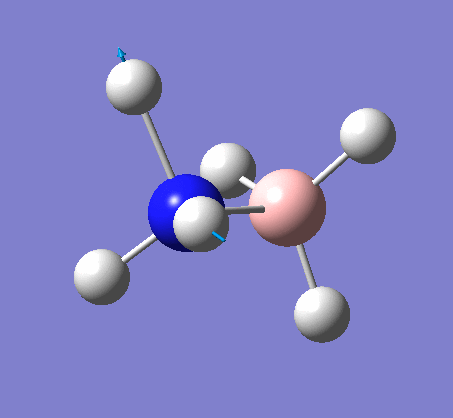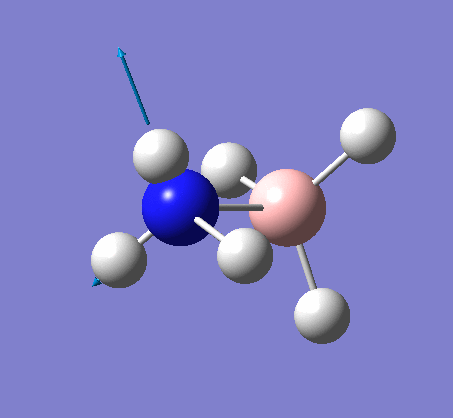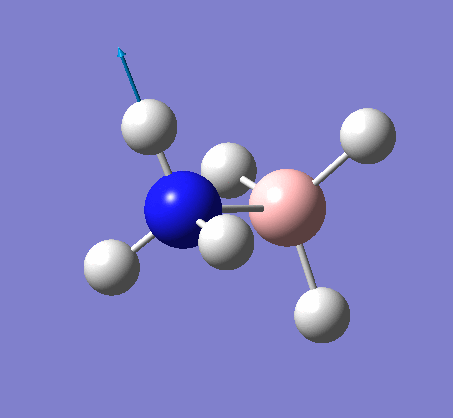
NH3
Optimization
Optimization was achieved using the DFT/B3LYP method and 6-31G(d,p) basis set.
This proceeded to give the following .log file: File:Rl nh3opt.log.

| N-H length | H-N-H angle |
|---|---|
 |

|
| 1.02Å | 105.7° |
The optimization was successful, and the following data shows that all the parameters converged:
Item Value Threshold Converged?
Maximum Force 0.000024 0.000450 YES
RMS Force 0.000012 0.000300 YES
Maximum Displacement 0.000088 0.001800 YES
RMS Displacement 0.000056 0.001200 YES
Predicted change in Energy=-1.756558D-09
Optimization completed.
-- Stationary point found.
----------------------------
! Optimized Parameters !
! (Angstroms and Degrees) !
-------------------------- --------------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------------------
! R1 R(1,2) 1.018 -DE/DX = 0.0 !
! R2 R(1,3) 1.018 -DE/DX = 0.0 !
! R3 R(1,4) 1.018 -DE/DX = 0.0 !
! A1 A(2,1,3) 105.7414 -DE/DX = 0.0 !
! A2 A(2,1,4) 105.7486 -DE/DX = 0.0 !
! A3 A(3,1,4) 105.7478 -DE/DX = 0.0 !
! D1 D(2,1,4,3) -111.8632 -DE/DX = 0.0 !
--------------------------------------------------------------------------------
GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
The data above shows that the optimized NH3 molecule has N-H bond lengths of 1.02Å, and H-N-H bond angles of 105.7°. The symmetry point group is C1, and has a 1.8464 Debye dipole moment as it is not highly symmetric (not trigonal planar like BH3). The job took 4.0 seconds to run. The point group is incorrect as the nitrogen has a lone pair. This occurs because the keyword nosymm allows gaussview to calculate without considering geometry. Again in order to ensure the minima was found, the frequency analysis must be run.
Frequency analysis
Again the same method was used, changing the job to frequency. This frequency analysis gave the following .log file: File:Rl nh3freq.log

| N-H length | H-N-H angle |
|---|---|
 |

|
| 1.02Å | 105.7° |
The optimization was successful, and the following data shows that all the parameters converged:
Item Value Threshold Converged?
Maximum Force 0.000125 0.000450 YES
RMS Force 0.000068 0.000300 YES
Maximum Displacement 0.000958 0.001800 YES
RMS Displacement 0.000581 0.001200 YES
Predicted change in Energy=-2.120560D-07
Optimization completed.
-- Stationary point found.
GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
Low frequencies --- 0.0002 0.0014 0.0014 17.1982 22.5909 39.0400 Low frequencies --- 265.9020 632.3790 639.0769
Vibrational analysis
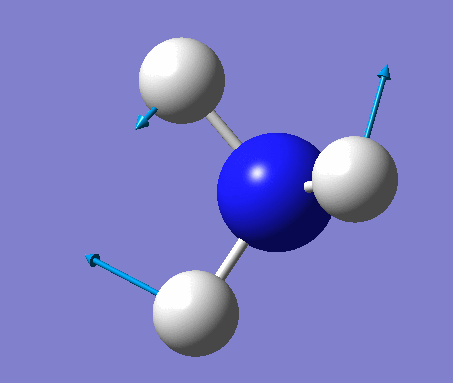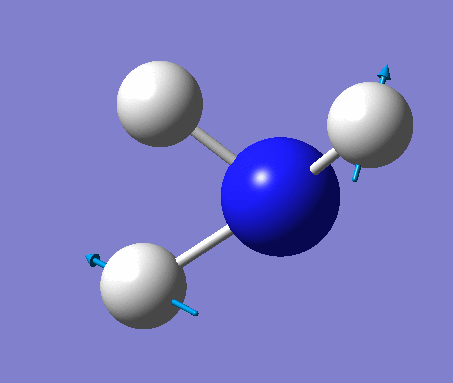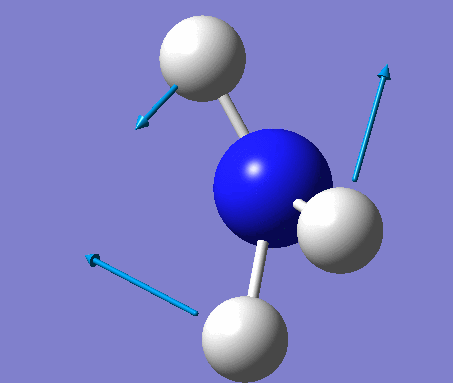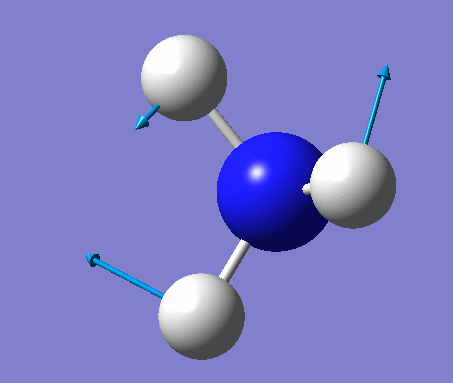
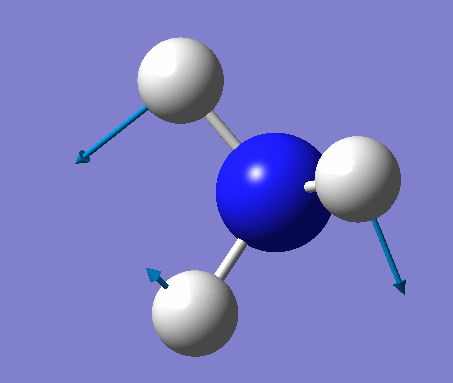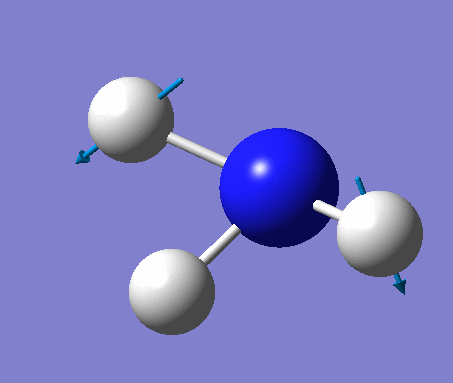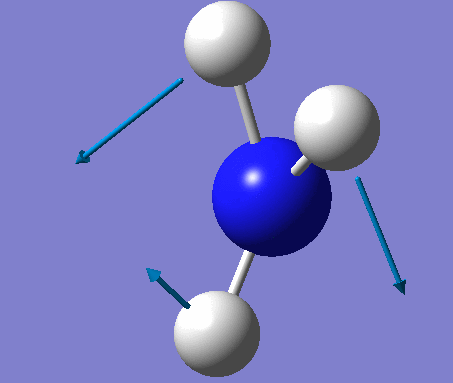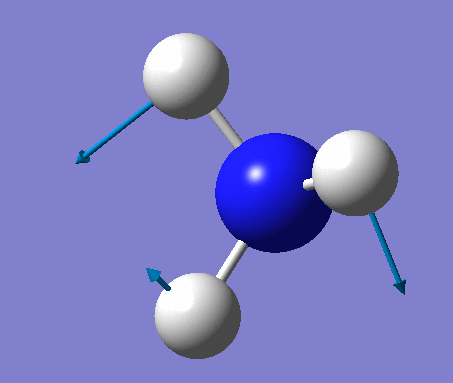
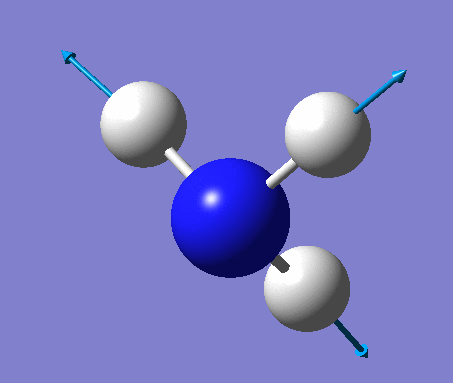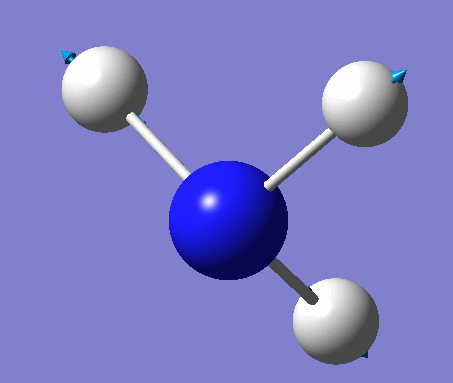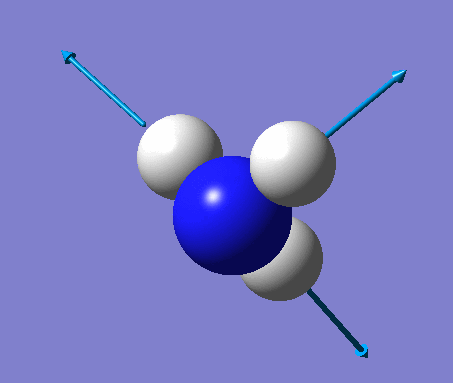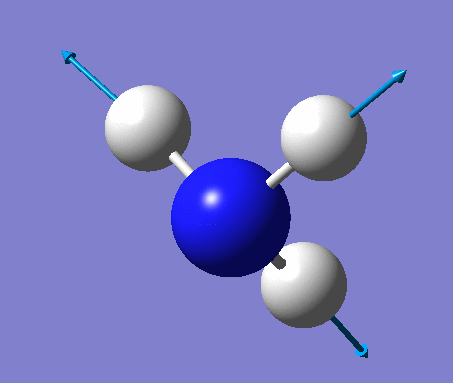
| No. | Vibration form | Frequency (cm-1) | Intensity | Symmetry (C1 point group) | Description |
|---|---|---|---|---|---|
| 1 | Wagging  |
1090 | 145 | A | All three H's move together in the same direction, with the N moving in the opposite direction at all times. |
| 2 | Scissoring  |
1694 | 14 | A | Two of the H's move down, while the other H and N move up to counteract this motion. Thus one H-N-H angle is contracted while the other two angles are expanded. |
| 3 | Rocking  |
1694 | 14 | A | One H moves a lot to contract one H-N-H angle and expand another. The third angle is held constant with the rest of the atoms moving to keep up with the moving H. |
| 4 | Symmetric stretching  |
3461 | 1 | A | Symmetrical stretching of all three H's at the same time. |
| 5 | Asymmetric stretching  |
3589 | 0.3 | A | One N-H bond stays the same length whilst the other two lengthen and shorten at the same time. |
| 6 | Asymmetric stretching  |
3590 | 0.3 | A | Two of the N-H bonds stretch symmetrically, whilst the third one stretches asymmetrically relative to the other two. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
This gives the corresponding IR spectrum:

Mode 1 causes the most significant change in dipole moment, which is shown by it having the most intense peak in the IR spectrum. Modes 2 and 3 are degenerate, meaning only one peak is shown. Modes 5 and 6 do not cause a significant change and thus has such a weak intensity that the peak does not show up on the spectrum.
Molecular orbital analysis
The same method was used, with the job set as energy. The .chk file was used to analyze the MOs, and the .log file was used to analyze the charge distribution. The analysis gave the following .log file: Media:Rl_nh3nbo.log
| MO | Image | Energy (a.u.) | MO | Image | Energy (a.u.) |
|---|---|---|---|---|---|
| 1 |  |
-14.30568 | 5 |  |
-0.25317 |
| 2 |  |
-0.84466 | 6 |  |
0.07985 |
| 3 |  |
-0.45030 | 7 |  |
0.16922 |
| 4 |  |
-0.45029 | 8 |  |
0.16923 |
These MOs are very similar to those for BH3. However due to its lone pair it has two more electrons in its MO diagram and thus has a different HOMO-LUMO. The energies of the MOs here would be lower than for BH3 since N is more electronegative and thus lowers the energy levels.
Charge distribution
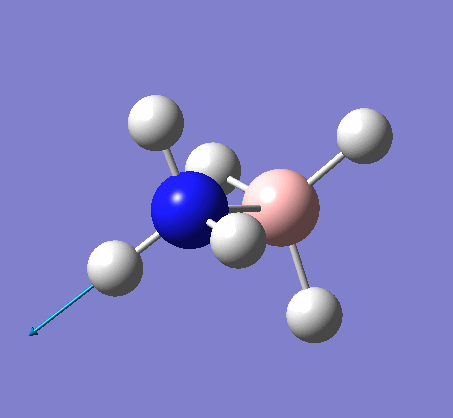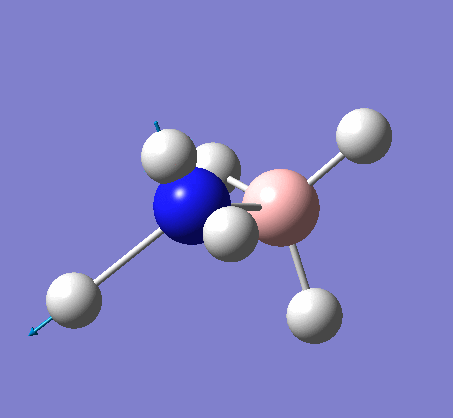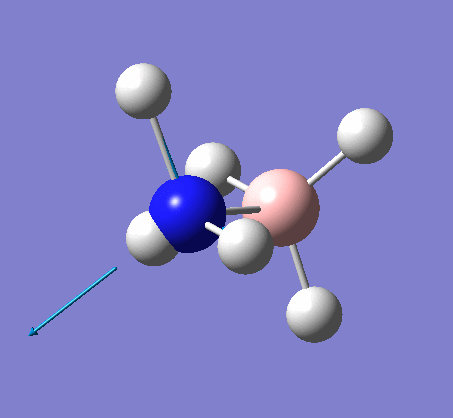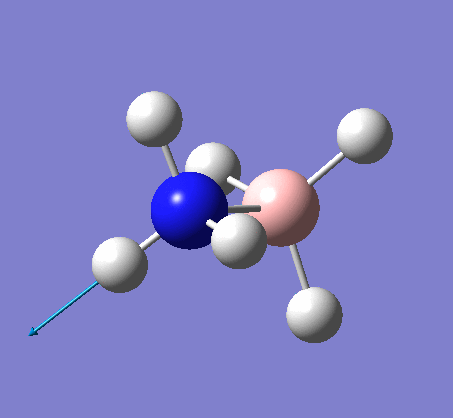
A NBO analysis was run on the .log file of the optimized NH3 molecule. The red color indicates a negatively charged region, and the green color a positively charged region. The color range was from -1.000 to +1.000.
The relative charges were +0.375 for all three H atoms, and -1.125 for the central N atom, as shown by the diagram to the left and below.


The following part of the .log file shows the natural charge information.
Summary of Natural Population Analysis:
Natural Population
Natural -----------------------------------------------
Atom No Charge Core Valence Rydberg Total
-----------------------------------------------------------------------
N 1 -1.12515 1.99982 6.11104 0.01429 8.12515
H 2 0.37505 0.00000 0.62250 0.00246 0.62495
H 3 0.37505 0.00000 0.62250 0.00246 0.62495
H 4 0.37505 0.00000 0.62249 0.00246 0.62495
=======================================================================
* Total * 0.00000 1.99982 7.97852 0.02166 10.00000
The following information from the .log file gives precise details on the electron density. Orbitals 1, 2, and 3 show that 68.83% of the N-H bond is contributed from N. Of this percentage, 24.87% is of s-orbital character, 75.05% of p-orbital character, and 0.09% of d-orbital character. This shows that the bond is an sp3 bond. Orbital 4 is of 100% on the N, and is of s-orbital character. Orbital 5 is a lone pair, giving a 1.99721 occupancy, which is close to 2 electrons. This shows that this orbital is formally occupied. This N-H bond is of 2c2e character.
(Occupancy) Bond orbital/ Coefficients/ Hybrids
---------------------------------------------------------------------------------
1. (1.99909) BD ( 1) N 1 - H 2
( 68.83%) 0.8297* N 1 s( 24.87%)p 3.02( 75.05%)d 0.00( 0.09%)
-0.0001 -0.4986 -0.0059 0.0000 -0.2910
0.0052 0.8155 0.0277 0.0000 0.0000
0.0281 0.0000 0.0000 0.0032 0.0082
( 31.17%) 0.5583* H 2 s( 99.91%)p 0.00( 0.09%)
-0.9996 0.0000 0.0072 -0.0289 0.0000
2. (1.99909) BD ( 1) N 1 - H 3
( 68.83%) 0.8297* N 1 s( 24.86%)p 3.02( 75.05%)d 0.00( 0.09%)
0.0001 0.4986 0.0059 0.0000 0.2910
-0.0052 0.4077 0.0138 0.7062 0.0240
0.0140 0.0243 0.0076 0.0033 0.0031
( 31.17%) 0.5583* H 3 s( 99.91%)p 0.00( 0.09%)
0.9996 0.0000 -0.0072 -0.0145 -0.0250
3. (1.99909) BD ( 1) N 1 - H 4
( 68.83%) 0.8297* N 1 s( 24.87%)p 3.02( 75.05%)d 0.00( 0.09%)
0.0001 0.4986 0.0059 0.0000 0.2909
-0.0052 0.4077 0.0138 -0.7062 -0.0239
0.0140 -0.0243 -0.0076 0.0033 0.0031
( 31.17%) 0.5583* H 4 s( 99.91%)p 0.00( 0.09%)
0.9996 0.0000 -0.0072 -0.0145 0.0250
4. (1.99982) CR ( 1) N 1 s(100.00%)
1.0000 -0.0002 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.0000 0.0000
5. (1.99721) LP ( 1) N 1 s( 25.38%)p 2.94( 74.52%)d 0.00( 0.10%)
0.0001 0.5036 -0.0120 0.0000 -0.8618
0.0505 0.0000 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 -0.0269 0.0155
The following information from the .log file shows that all the N-H bonds have the same occupancy and energy, whilst the core s-orbital character orbital (4) has a significantly lower energy. The lone pair has a higher energy.
Natural Bond Orbitals (Summary):
Principal Delocalizations
NBO Occupancy Energy (geminal,vicinal,remote)
====================================================================================
Molecular unit 1 (H3N)
1. BD ( 1) N 1 - H 2 1.99909 -0.60417
2. BD ( 1) N 1 - H 3 1.99909 -0.60417
3. BD ( 1) N 1 - H 4 1.99909 -0.60416
4. CR ( 1) N 1 1.99982 -14.16768
5. LP ( 1) N 1 1.99721 -0.31756
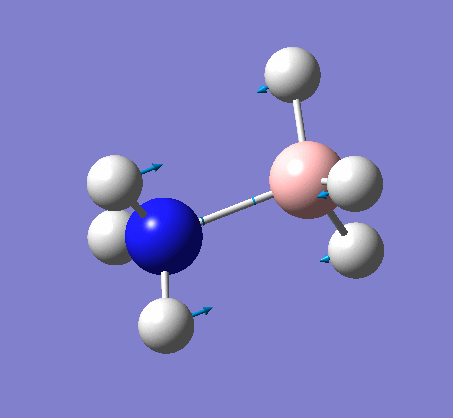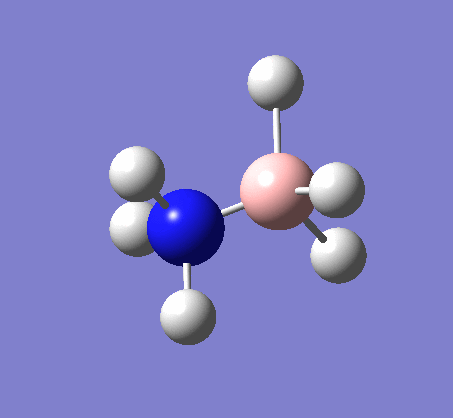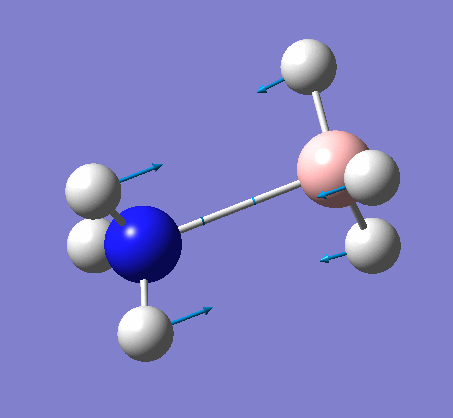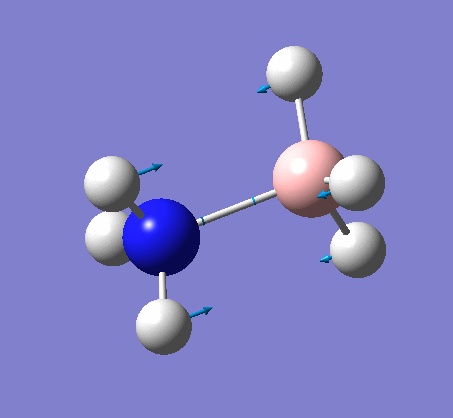
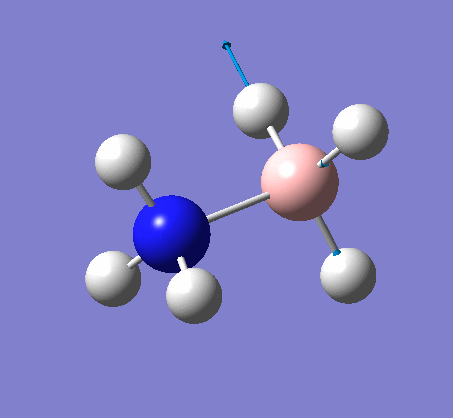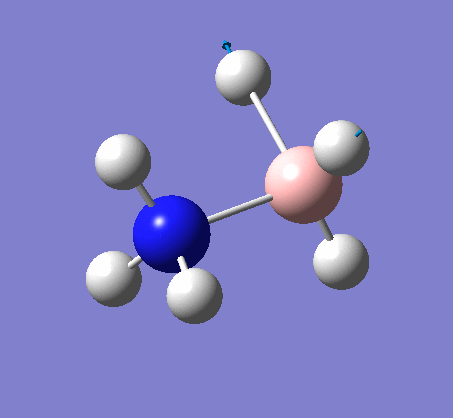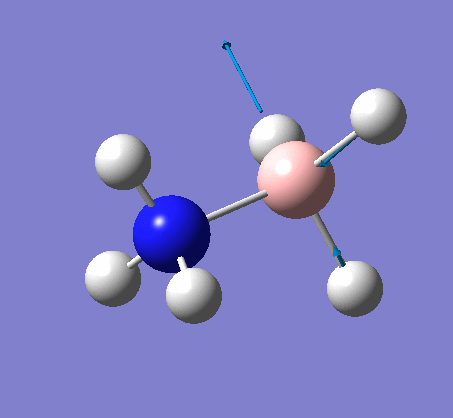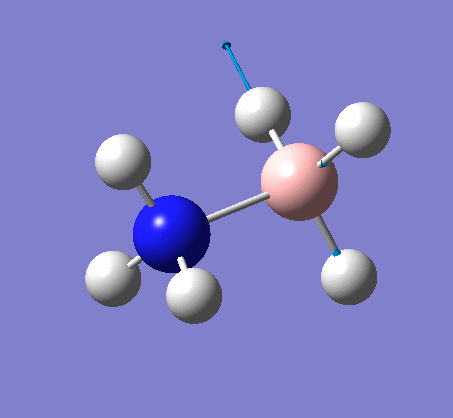
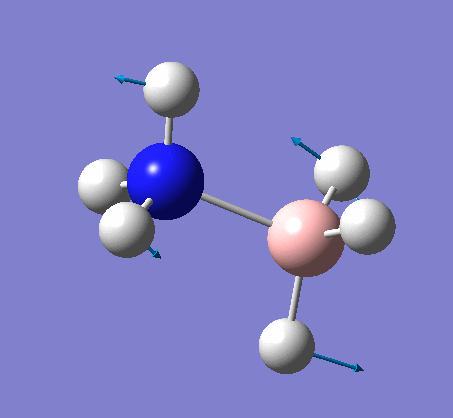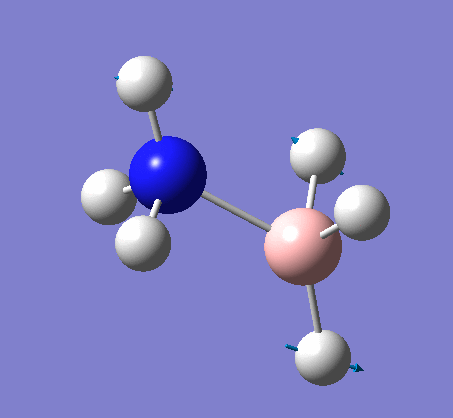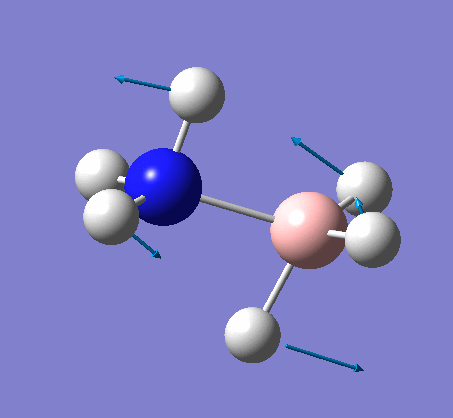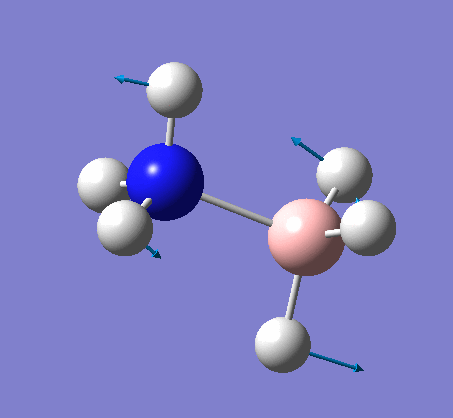
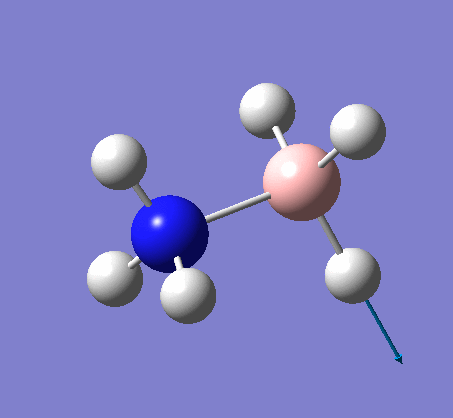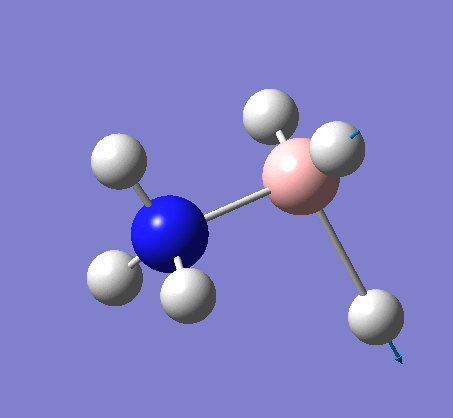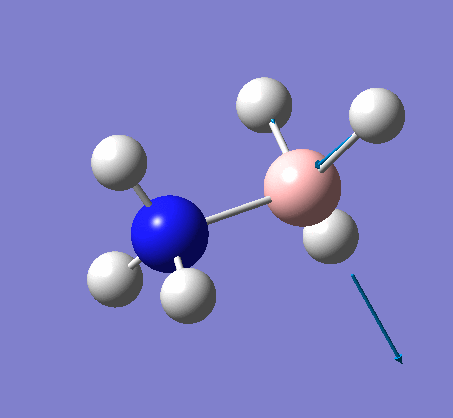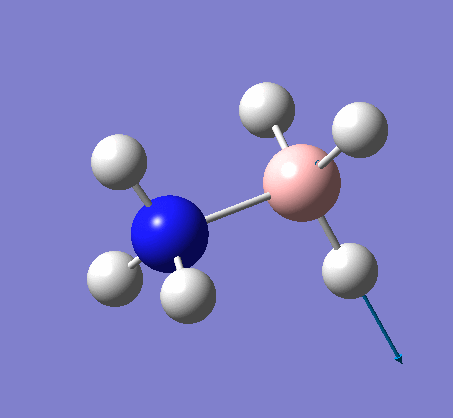
NH3BH3
Optimization
Optimization was achieved using the DFT/B3LYP method and 6-31G(d,p) basis set.
This proceeded to give the following .log file: File:Rl nh3bh3opt.log.

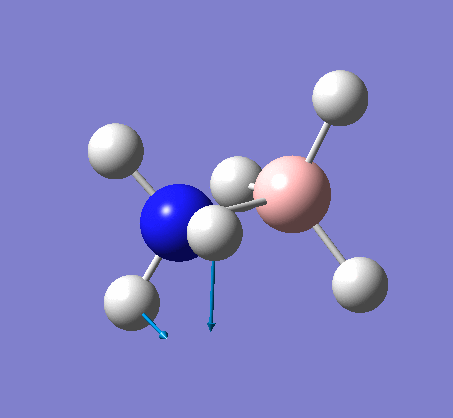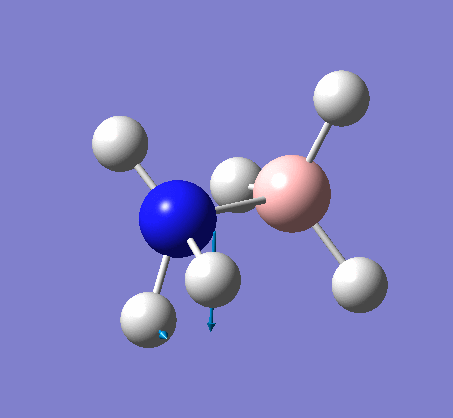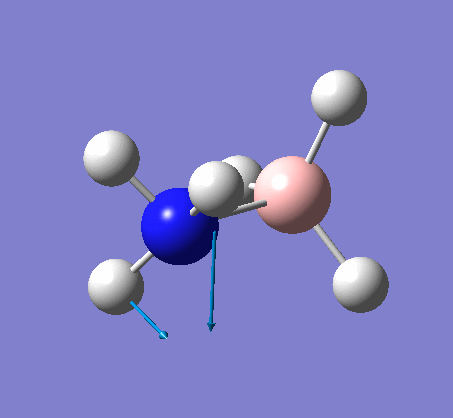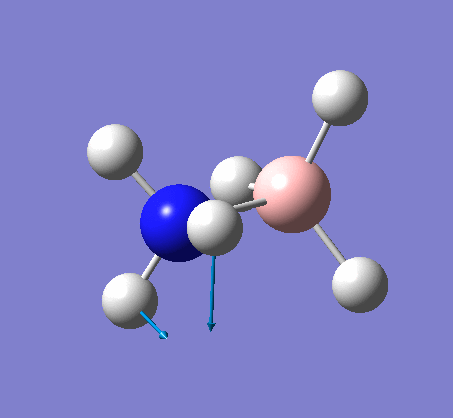
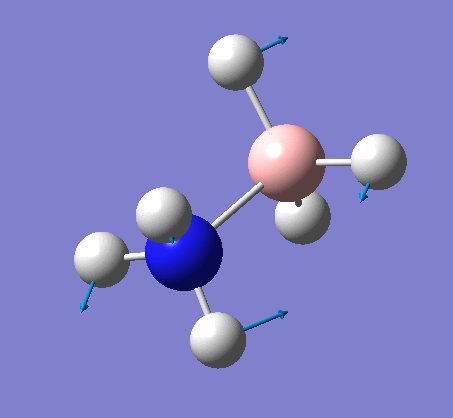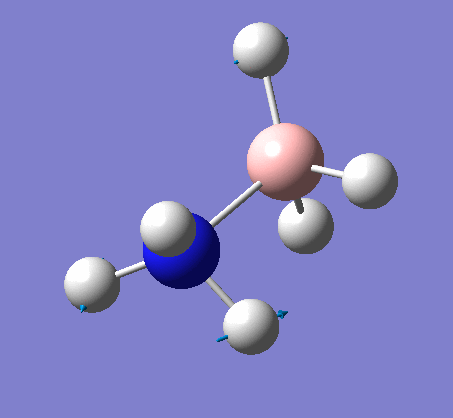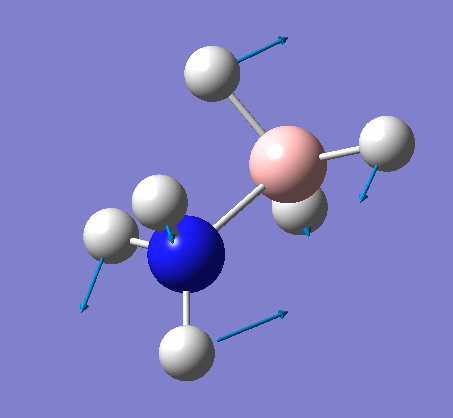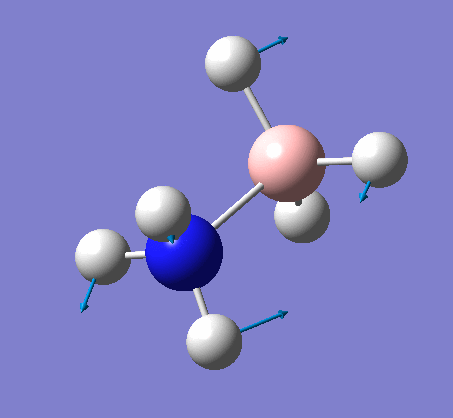
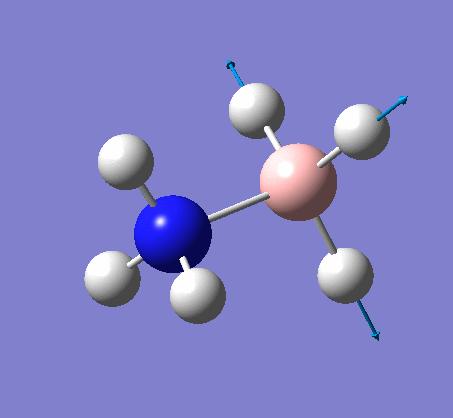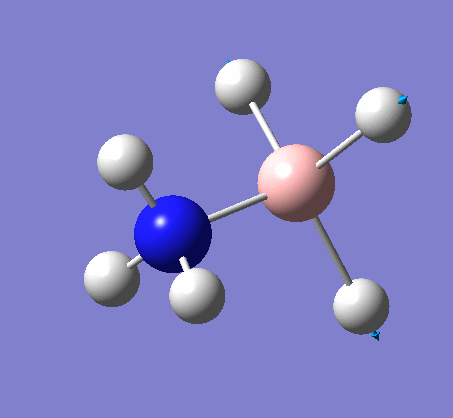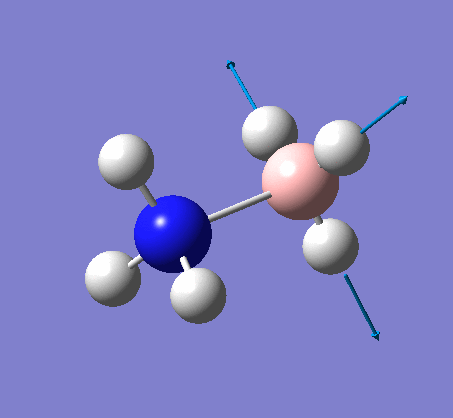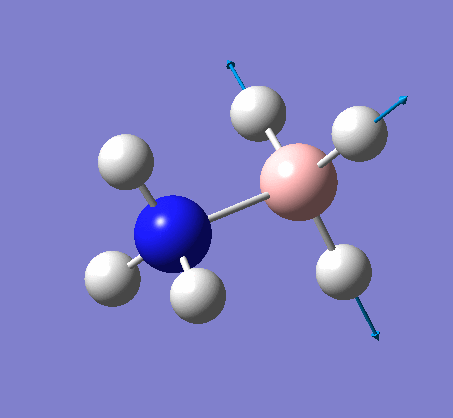
| X-H length | H-X-H angle | |
|---|---|---|
| X = N |  |

|
| 1.02Å | 107.8° | |
| X = B |  |

|
| 1.21Å | 113.9° |
The central N-B bond length is 1.67Å.
The optimization was successful, and the following data shows that all the parameters converged:
Item Value Threshold Converged?
Maximum Force 0.000138 0.000450 YES
RMS Force 0.000063 0.000300 YES
Maximum Displacement 0.000755 0.001800 YES
RMS Displacement 0.000462 0.001200 YES
Predicted change in Energy=-2.046278D-07
Optimization completed.
-- Stationary point found.
----------------------------
! Optimized Parameters !
! (Angstroms and Degrees) !
-------------------------- --------------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------------------
! R1 R(1,7) 1.0186 -DE/DX = -0.0001 !
! R2 R(2,7) 1.0186 -DE/DX = -0.0001 !
! R3 R(3,7) 1.0186 -DE/DX = -0.0001 !
! R4 R(4,8) 1.2101 -DE/DX = -0.0001 !
! R5 R(5,8) 1.2101 -DE/DX = -0.0001 !
! R6 R(6,8) 1.2101 -DE/DX = -0.0001 !
! R7 R(7,8) 1.668 -DE/DX = -0.0001 !
! A1 A(1,7,2) 107.8699 -DE/DX = 0.0 !
! A2 A(1,7,3) 107.8652 -DE/DX = 0.0 !
! A3 A(1,7,8) 111.0329 -DE/DX = 0.0 !
! A4 A(2,7,3) 107.8696 -DE/DX = 0.0 !
! A5 A(2,7,8) 111.0286 -DE/DX = 0.0 !
! A6 A(3,7,8) 111.0291 -DE/DX = 0.0 !
! A7 A(4,8,5) 113.8694 -DE/DX = 0.0 !
! A8 A(4,8,6) 113.8722 -DE/DX = 0.0 !
! A9 A(4,8,7) 104.6003 -DE/DX = 0.0 !
! A10 A(5,8,6) 113.8747 -DE/DX = 0.0 !
! A11 A(5,8,7) 104.6003 -DE/DX = 0.0 !
! A12 A(6,8,7) 104.5984 -DE/DX = 0.0 !
! D1 D(1,7,8,4) 180.0133 -DE/DX = 0.0 !
! D2 D(1,7,8,5) -59.9892 -DE/DX = 0.0 !
! D3 D(1,7,8,6) 60.0135 -DE/DX = 0.0 !
! D4 D(2,7,8,4) -59.9839 -DE/DX = 0.0 !
! D5 D(2,7,8,5) 60.0136 -DE/DX = 0.0 !
! D6 D(2,7,8,6) 180.0163 -DE/DX = 0.0 !
! D7 D(3,7,8,4) 60.0161 -DE/DX = 0.0 !
! D8 D(3,7,8,5) 180.0136 -DE/DX = 0.0 !
! D9 D(3,7,8,6) -59.9837 -DE/DX = 0.0 !
--------------------------------------------------------------------------------
GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
The data above shows that the optimized NH3BH3 molecule has N-H bond lengths of 1.02Å, H-N-H bond angles of 107.8°, B-H bond lengths of 1.21Å, H-B-H bond angles of 113.9°, and a central N-B bond length of 1.67Å. The symmetry group is C1 and has a 5.5654 Debye dipole moment as it is not highly symmetric. The frequency analysis is run next to determine if the minima was found.
Frequency analysis
The same method was used, with job = frequency. This frequency analysis gave the following .log file: File:Rl nh3freq.log

| X-H length | H-X-H angle | |
|---|---|---|
| X = N |  |

|
| 1.02Å | 107.9° | |
| X = B |  |

|
| 1.21Å | 113.9° |
The optimization was successful, and the following data shows that all the parameters converged:
Item Value Threshold Converged?
Maximum Force 0.000022 0.000450 YES
RMS Force 0.000009 0.000300 YES
Maximum Displacement 0.000078 0.001800 YES
RMS Displacement 0.000039 0.001200 YES
Predicted change in Energy=-1.599836D-09
Optimization completed.
-- Stationary point found.
GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
Low frequencies --- -30.7927 -0.0015 0.0005 0.0011 20.2690 28.2324 Low frequencies --- 1089.5544 1694.1237 1694.1863
Vibrational analysis
| No. | Vibration form | Frequency (cm-1) | Literature frequency value (cm-1) [6] | Intensity | Symmetry (C1 point group) | Description | No. | Vibration form | Frequency (cm-1) | Literature frequency value (cm-1) | Intensity | Symmetry (C1 point group) | Description |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Wagging about B-N bond  |
266 | not observed | 0.0000 | A | The molecule back and forth on both ends around the B-N bond. | 10 | NH3 wagging  |
1330 | 1343 | 114 | A | The three H's on the NH3 end move back and forth. |
| 2 | B-N stretching  |
632 | 603 | 14 | A | Both ends of the molecule move back and forth resulting in B-N stretching. | 11 | NH3 scissoring  |
1676 | 1608 | 28 | A | One H-N-H angle is constricted whilst the other two are expanded. |
| 3 | B-N bending  |
639 | not reported | 4 | A | Two H's on each end move back and forth causing the B-N bond to bend. | 12 | NH3 rocking  |
1676 | 1608 | 28 | A | One H-N-H angle is held constant, one is expanded, and the other is constricted. |
| 4 | B-N bending  |
640 | not reported | 4 | A | On each end, one H moves in one direction while the other two move in the other. This causes the B-N to bend (more so than in mode 3). | 13 | BH3 symmetric stretching  |
2470 | 2340 | 67 | A | All three H's on the B side stretch in a concerted fashion. |
| 5 | B-N seesaw  |
1069 | 968 | 40 | A | Two H's on each end move back and forth in a way that the B-N bond seesaws. | 14 | BH3 asymmetric stretching  |
2530 | 2415 | 231 | A | One B-H length stays constant whilst the other two lengthen and shorten at the same time. |
| 6 | B-N seesaw  |
1060 | 987 | 40 | A | On each end, one H moves in one direction while the other two move in the other in such a way that the B-N bond seesaws (more than in mode 5). | 15 | BH3 asymmetric stretching  |
2530 | 2415 | 231 | A | Same as in mode 14. |
| 7 | BH3 wagging  |
1197 | 1055 | 109 | A | The three H's on the BH3 end move back and forth. | 16 | NH3 symmetric stretching  |
3462 | 3337 | 3 | A | All three H's on the N side stretch in a concerted fashion. |
| 8 | BH3 scissoring  |
1204 | 1216 | 3 | A | Two of the H-B-H angles are expanded whilst the third is constricted. | 17 | NH3 asymmetric stretching  |
3579 | 3386 | 28 | A | One N-H length stays constant whilst the other two lengthen and shorten at the same time. |
| 9 | BH3 rocking  |
1204 | 1216 | 3 | A | One H-B-H angle is held constant, while another is being expanded, and the third is being constricted. | 18 | NH3 asymmetric stretching  |
3579 | 3386 | 28 | A | Same as in mode 17. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
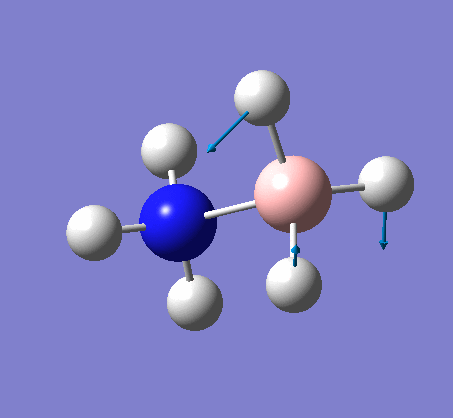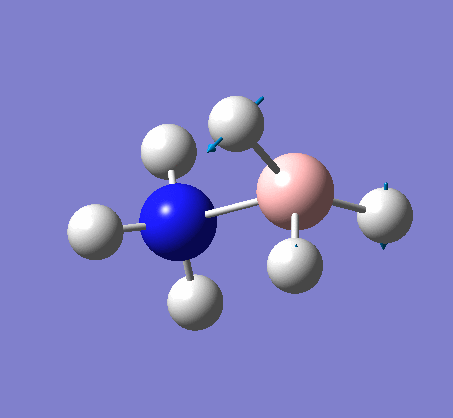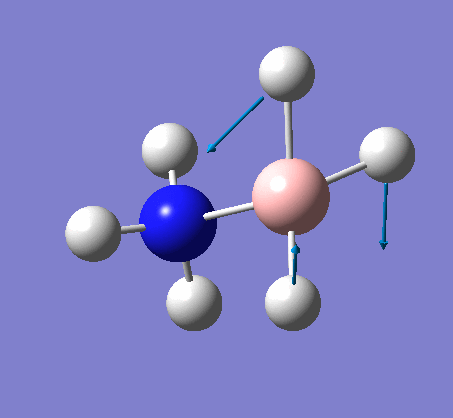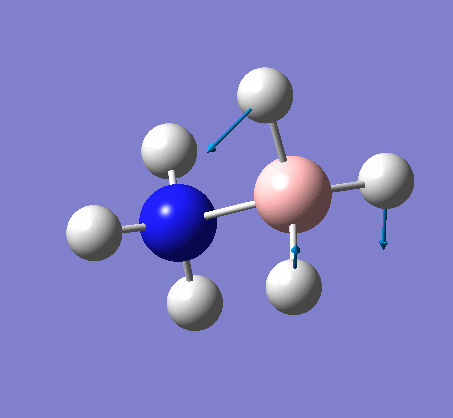{kind=link}
{kind=link}
This gives the corresponding IR spectrum:

Association energy
E(BH3) = -26.61532252 a.u.
E(NH3) = -56.55776856 a.u.
E(NH3BH3) = -83.22468919 a.u.
ΔE = E(NH3BH3) - [E(BH3) + E(NH3)]
ΔE = -83.22468919 a.u. - [(-26.61532252 a.u.) + (-56.55776856 a.u.)]
ΔE = -0.05159811 a.u.
1 a.u. = 2625.50184 kJ/mol
Thus, ΔE = -135.47 kJ/mol
This value is a bit lower than that found experimentally: -172.1 kJ/mol [7]
Mini project - Aromaticity
An investigation of the MOs and energies of benzene and its isoelectronic analogues: boratabenzene, pyridinium, and borazine. All calculations were performed on the HPC server and the files published to D-space.
Benzene
Optimization
The molecule was optimized using the DFT/B3LYP method and 6-31G(d,p) basis set. This file was submitted to D-space: [6]

| C-H length | H-C-C angle |
|---|---|
 |

|
| 1.09Å | 120.0° |
The C-C bond length is 1.39Å.
The following information from the .log file shows that the parameters converged:
Item Value Threshold Converged?
Maximum Force 0.000212 0.000450 YES
RMS Force 0.000085 0.000300 YES
Maximum Displacement 0.000991 0.001800 YES
RMS Displacement 0.000315 0.001200 YES
Predicted change in Energy=-5.157454D-07
Optimization completed.
-- Stationary point found.
----------------------------
! Optimized Parameters !
! (Angstroms and Degrees) !
-------------------------- --------------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------------------
! R1 R(1,2) 1.3963 -DE/DX = 0.0001 !
! R2 R(1,6) 1.3961 -DE/DX = 0.0002 !
! R3 R(1,7) 1.0861 -DE/DX = 0.0002 !
! R4 R(2,3) 1.3961 -DE/DX = 0.0002 !
! R5 R(2,8) 1.0861 -DE/DX = 0.0002 !
! R6 R(3,4) 1.3963 -DE/DX = 0.0001 !
! R7 R(3,9) 1.086 -DE/DX = 0.0002 !
! R8 R(4,5) 1.3961 -DE/DX = 0.0002 !
! R9 R(4,10) 1.086 -DE/DX = 0.0002 !
! R10 R(5,6) 1.3963 -DE/DX = 0.0001 !
! R11 R(5,11) 1.0861 -DE/DX = 0.0002 !
! R12 R(6,12) 1.0861 -DE/DX = 0.0002 !
! A1 A(2,1,6) 119.9972 -DE/DX = 0.0 !
! A2 A(2,1,7) 119.9949 -DE/DX = 0.0 !
! A3 A(6,1,7) 120.0079 -DE/DX = 0.0 !
! A4 A(1,2,3) 120.0079 -DE/DX = 0.0 !
! A5 A(1,2,8) 119.9881 -DE/DX = 0.0 !
! A6 A(3,2,8) 120.004 -DE/DX = 0.0 !
! A7 A(2,3,4) 119.9948 -DE/DX = 0.0 !
! A8 A(2,3,9) 120.0086 -DE/DX = 0.0 !
! A9 A(4,3,9) 119.9966 -DE/DX = 0.0 !
! A10 A(3,4,5) 119.9972 -DE/DX = 0.0 !
! A11 A(3,4,10) 119.9934 -DE/DX = 0.0 !
! A12 A(5,4,10) 120.0094 -DE/DX = 0.0 !
! A13 A(4,5,6) 120.0083 -DE/DX = 0.0 !
! A14 A(4,5,11) 120.0014 -DE/DX = 0.0 !
! A15 A(6,5,11) 119.9904 -DE/DX = 0.0 !
! A16 A(1,6,5) 119.9946 -DE/DX = 0.0 !
! A17 A(1,6,12) 120.0106 -DE/DX = 0.0 !
! A18 A(5,6,12) 119.9948 -DE/DX = 0.0 !
! D1 D(6,1,2,3) -0.0059 -DE/DX = 0.0 !
! D2 D(6,1,2,8) 180.0023 -DE/DX = 0.0 !
! D3 D(7,1,2,3) -180.01 -DE/DX = 0.0 !
! D4 D(7,1,2,8) -0.0019 -DE/DX = 0.0 !
! D5 D(2,1,6,5) -0.0055 -DE/DX = 0.0 !
! D6 D(2,1,6,12) -179.9972 -DE/DX = 0.0 !
! D7 D(7,1,6,5) -180.0013 -DE/DX = 0.0 !
! D8 D(7,1,6,12) 0.007 -DE/DX = 0.0 !
! D9 D(1,2,3,4) 0.0117 -DE/DX = 0.0 !
! D10 D(1,2,3,9) -179.9914 -DE/DX = 0.0 !
! D11 D(8,2,3,4) 180.0036 -DE/DX = 0.0 !
! D12 D(8,2,3,9) 0.0005 -DE/DX = 0.0 !
! D13 D(2,3,4,5) -0.0062 -DE/DX = 0.0 !
! D14 D(2,3,4,10) -180.0059 -DE/DX = 0.0 !
! D15 D(9,3,4,5) 179.9969 -DE/DX = 0.0 !
! D16 D(9,3,4,10) -0.0028 -DE/DX = 0.0 !
! D17 D(3,4,5,6) -0.0051 -DE/DX = 0.0 !
! D18 D(3,4,5,11) 180.0058 -DE/DX = 0.0 !
! D19 D(10,4,5,6) -180.0055 -DE/DX = 0.0 !
! D20 D(10,4,5,11) 0.0054 -DE/DX = 0.0 !
! D21 D(4,5,6,1) 0.011 -DE/DX = 0.0 !
! D22 D(4,5,6,12) 180.0027 -DE/DX = 0.0 !
! D23 D(11,5,6,1) -179.9999 -DE/DX = 0.0 !
! D24 D(11,5,6,12) -0.0082 -DE/DX = 0.0 !
--------------------------------------------------------------------------------
GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
The data above shows that the optimized benzene molecule has C-H bond lengths of 1.09Å, H-B-H bond angles of 120.0°, and C-C bond lengths of 1.39Å. The symmetry point group is C1, and has a 0.00 Debye dipole moment due to its high symmetry. The final energy is -232.25820551 a.u., and took 2 min 3.1 seconds to run.
Frequency
This frequency analysis was submitted to D-space: [[7]]

| C-H length | H-C-C angle |
|---|---|
 |

|
| 1.09Å | 120.0° |
The frequency calculation was successful, and the following data shows that all the parameters converged:
Item Value Threshold Converged?
Maximum Force 0.000195 0.000450 YES
RMS Force 0.000095 0.000300 YES
Maximum Displacement 0.001064 0.001800 YES
RMS Displacement 0.000381 0.001200 YES
Predicted change in Energy=-5.065853D-07
Optimization completed.
-- Stationary point found.
GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
Low frequencies --- -17.2788 -14.5868 -9.6527 -0.0013 -0.0011 -0.0009 Low frequencies --- 413.7971 414.4697 620.8545
Vibrational analysis
| No. | Frequency (cm-1) | Intensity | Symmetry (C1 point group) | Description |
|---|---|---|---|---|
| 1 | 414 | 0 | A | One pair of opposite C-H wagging in and out of the plane. |
| 2 | 414 | 0 | A | The other 2 pairs of opposite C-H wagging in and out of the plane. |
| 3 | 621 | 0 | A | C-C bending in plane in the same direction. |
| 4 | 621 | 0 | A | C-C bending in plane in opposite directions. |
| 5 | 693 | 74 | A | Simultaneous H wagging in and out of the plane in the same direction. |
| 6 | 718 | 0 | A | C-C in and out of plane see-sawing. |
| 7 | 864 | 0 | A | All three pairs of opposite H's concerted wagging in and out of the plane. |
| 8 | 864 | 0 | A | Two pairs of opposite H's concerted wagging in and out of the plane. |
| 9 | 974 | 0 | A | Opposite H pairs alternate wagging in and out of the plane. |
| 10 | 974 | 0 | A | Opposite H pairs alternate wagging in and out of the plane. |
| 11 | 1013 | 0 | A | Opposite H pairs alternate wagging in and out of the plane. |
| 12 | 1018 | 0 | A | Alternate C-C in plane asymmetric stretching |
| 13 | 1020 | 0 | A | C-C in plane symmetric stretching |
| 14 | 1066 | 3 | A | C-C in plane asymmetric stretching |
| 15 | 1066 | 3 | A | C-C in plane asymmetric stretching |
| 16 | 1179 | 0 | A | C-H in plane asymmetric bending. |
| 17 | 1202 | 0 | A | Two pairs opposite C-H in plane symmetric bending, other pair asymmetric bending. |
| 18 | 1202 | 0 | A | C-H in plane asymmetric bending. |
| 19 | 1356 | 0 | A | C-H in plane asymmetric stretching. |
| 20 | 1380 | 0 | A | C-H in plane symmetric bending. |
| 21 | 1524 | 7 | A | C-C in plane asymmetric stretching with C-H in plane asymmetric bending. |
| 22 | 1524 | 7 | A | C-C in plane asymmetric stretching with C-H in plane asymmetric bending. |
| 23 | 1653 | 0 | A | C-C in plane asymmetric stretching. |
| 24 | 1653 | 0 | A | C-C in plane asymmetric stretching. |
| 25 | 3175 | 0 | A | Alternate C-H in plane stretching. |
| 26 | 3185 | 0 | A | C-H in plane asymmetric stretching. |
| 27 | 3185 | 0 | A | C-H in plane asymmetric stretching. |
| 28 | 3200 | 47 | A | Opposite C-H groups in plane asymmetric stretching. |
| 29 | 3200 | 47 | A | Two pairs of C-H groups in plane asymmetric stretching. |
| 30 | 3211 | 0 | A | C-H symmetric stretching. |
Most of these modes result in no change in dipole moment and thus do not produce peaks in the spectrum. Only modes 5, 14 and 15 (degenerate pair), 21 and 22 (degenerate pair), and 28 and 29 (degenerate pair) produce peaks intense enough to be observed.
This gives the following IR spectrum:

MO analysis
The same method was used here, whilst changing the job to energy. The MO analysis submitted to D-space link is: [[8]]
The first 6 MOs of benzene are part of the core molecular orbitals as they involve the 2s orbitals of the carbons. This does not affect the stability as much as MOs 7-23.
The "center" part of the MO diagram is as follows:

Charge Distribution
An NBO analysis was run on the .log file of the optimized benzene molecule. The red color indicates a negatively charged region, and the green color a positively charged region. The color range was from -0.500 to +0.500.


The relative charges were +0.239 for all H, -0.239 for all C.
The following part of the .log file shows the natural charge information:
Summary of Natural Population Analysis:
Natural Population
Natural -----------------------------------------------
Atom No Charge Core Valence Rydberg Total
-----------------------------------------------------------------------
C 1 -0.23852 1.99910 4.22611 0.01331 6.23852
C 2 -0.23855 1.99910 4.22613 0.01331 6.23855
C 3 -0.23854 1.99910 4.22613 0.01331 6.23854
C 4 -0.23852 1.99910 4.22611 0.01331 6.23852
C 5 -0.23855 1.99910 4.22613 0.01331 6.23855
C 6 -0.23854 1.99910 4.22613 0.01331 6.23854
H 7 0.23854 0.00000 0.76003 0.00144 0.76146
H 8 0.23853 0.00000 0.76003 0.00144 0.76147
H 9 0.23854 0.00000 0.76002 0.00144 0.76146
H 10 0.23854 0.00000 0.76003 0.00144 0.76146
H 11 0.23853 0.00000 0.76003 0.00144 0.76147
H 12 0.23854 0.00000 0.76002 0.00144 0.76146
=======================================================================
* Total * 0.00000 11.99462 29.91690 0.08847 42.00000
The following information from the .log file (showing only MO 7-23) gives precise details on electron density. It gives key information. For example, for MO 7 62.04% of the C-H bond is contributed from C, and of this percentage 29.57% is of s-orbital character, 70.39% p-orbital character, and 0.04% d-orbital character. This is the way to analyze each MO.
(Occupancy) Bond orbital/ Coefficients/ Hybrids
---------------------------------------------------------------------------------
7. (1.98305) BD ( 1) C 2 - H 8
( 62.04%) 0.7876* C 2 s( 29.57%)p 2.38( 70.39%)d 0.00( 0.04%)
0.0003 -0.5436 -0.0126 0.0010 0.1529
-0.0027 0.8248 -0.0144 0.0000 0.0000
-0.0060 0.0000 0.0000 0.0155 0.0105
( 37.96%) 0.6161* H 8 s( 99.95%)p 0.00( 0.05%)
-0.9997 -0.0014 -0.0042 -0.0224 0.0000
8. (1.98096) BD ( 1) C 3 - C 4
( 50.00%) 0.7071* C 3 s( 35.19%)p 1.84( 64.77%)d 0.00( 0.04%)
-0.0001 0.5932 -0.0078 0.0006 0.1263
-0.0282 0.7940 0.0218 0.0000 0.0000
0.0073 0.0000 0.0000 -0.0149 -0.0109
( 50.00%) 0.7071* C 4 s( 35.19%)p 1.84( 64.77%)d 0.00( 0.04%)
-0.0001 0.5932 -0.0078 0.0006 -0.1668
-0.0342 -0.7865 -0.0103 0.0000 0.0000
0.0045 0.0000 0.0000 -0.0160 -0.0109
9. (1.98305) BD ( 1) C 3 - H 9
( 62.04%) 0.7876* C 3 s( 29.58%)p 2.38( 70.38%)d 0.00( 0.04%)
0.0003 -0.5437 -0.0126 0.0010 0.7908
-0.0138 0.2799 -0.0049 -0.0001 0.0000
-0.0105 0.0000 0.0000 -0.0129 0.0105
( 37.96%) 0.6161* H 9 s( 99.95%)p 0.00( 0.05%)
-0.9997 -0.0014 -0.0215 -0.0076 0.0000
10. (1.98098) BD ( 1) C 4 - C 5
( 50.00%) 0.7071* C 4 s( 35.20%)p 1.84( 64.76%)d 0.00( 0.04%)
-0.0001 0.5933 -0.0079 0.0006 0.7508
0.0048 0.2875 0.0354 -0.0001 0.0000
0.0092 0.0000 0.0000 0.0138 -0.0109
( 50.00%) 0.7071* C 5 s( 35.20%)p 1.84( 64.76%)d 0.00( 0.04%)
-0.0001 0.5933 -0.0079 0.0006 -0.7645
-0.0260 -0.2487 0.0245 0.0000 0.0000
0.0116 0.0000 0.0000 0.0119 -0.0109
11. (1.66533) BD ( 2) C 4 - C 5
( 50.00%) 0.7071* C 4 s( 0.00%)p 1.00( 99.96%)d 0.00( 0.04%)
0.0000 0.0000 0.0000 0.0000 0.0000
0.0000 0.0001 0.0000 0.9997 -0.0133
0.0000 0.0191 -0.0037 0.0000 0.0000
( 50.00%) 0.7071* C 5 s( 0.00%)p 1.00( 99.96%)d 0.00( 0.04%)
0.0000 0.0000 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.9997 -0.0133
0.0000 -0.0125 -0.0149 0.0000 0.0000
12. (1.98305) BD ( 1) C 4 - H 10
( 62.04%) 0.7876* C 4 s( 29.58%)p 2.38( 70.39%)d 0.00( 0.04%)
0.0003 -0.5437 -0.0126 0.0010 0.6378
-0.0111 -0.5449 0.0095 0.0000 0.0000
0.0164 0.0000 0.0000 -0.0026 0.0105
( 37.96%) 0.6161* H 10 s( 99.95%)p 0.00( 0.05%)
-0.9997 -0.0014 -0.0173 0.0148 0.0000
13. (1.98096) BD ( 1) C 5 - C 6
( 50.00%) 0.7071* C 5 s( 35.20%)p 1.84( 64.76%)d 0.00( 0.04%)
-0.0001 0.5932 -0.0078 0.0006 0.6244
0.0330 -0.5064 0.0135 0.0001 0.0000
-0.0165 0.0000 0.0000 0.0011 -0.0109
( 50.00%) 0.7071* C 6 s( 35.19%)p 1.84( 64.77%)d 0.00( 0.04%)
-0.0001 0.5932 -0.0078 0.0006 -0.5978
0.0082 0.5377 0.0347 -0.0001 0.0000
-0.0161 0.0000 0.0000 0.0041 -0.0109
14. (1.98305) BD ( 1) C 5 - H 11
( 62.04%) 0.7876* C 5 s( 29.57%)p 2.38( 70.39%)d 0.00( 0.04%)
-0.0003 0.5436 0.0126 -0.0010 0.1530
-0.0027 0.8248 -0.0143 0.0000 0.0000
0.0060 0.0000 0.0000 -0.0155 -0.0105
( 37.96%) 0.6161* H 11 s( 99.95%)p 0.00( 0.05%)
0.9997 0.0014 -0.0042 -0.0224 0.0000
15. (1.98305) BD ( 1) C 6 - H 12
( 62.04%) 0.7876* C 6 s( 29.58%)p 2.38( 70.38%)d 0.00( 0.04%)
-0.0003 0.5437 0.0126 -0.0010 0.7907
-0.0138 0.2800 -0.0049 0.0000 0.0000
0.0105 0.0000 0.0000 0.0129 -0.0105
( 37.96%) 0.6161* H 12 s( 99.95%)p 0.00( 0.05%)
0.9997 0.0014 -0.0215 -0.0076 0.0000
16. (1.99911) CR ( 1) C 1 s(100.00%)p 0.00( 0.00%)
1.0000 0.0002 0.0000 0.0000 0.0002
0.0000 -0.0001 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.0000 0.0000
17. (1.99911) CR ( 1) C 2 s(100.00%)p 0.00( 0.00%)
1.0000 0.0002 0.0000 0.0000 0.0000
0.0000 -0.0002 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.0000 0.0000
18. (1.99911) CR ( 1) C 3 s(100.00%)p 0.00( 0.00%)
1.0000 0.0002 0.0000 0.0000 -0.0002
0.0000 -0.0001 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.0000 0.0000
19. (1.99911) CR ( 1) C 4 s(100.00%)p 0.00( 0.00%)
1.0000 0.0002 0.0000 0.0000 -0.0002
0.0000 0.0001 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.0000 0.0000
20. (1.99911) CR ( 1) C 5 s(100.00%)p 0.00( 0.00%)
1.0000 0.0002 0.0000 0.0000 0.0000
0.0000 0.0002 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.0000 0.0000
21. (1.99911) CR ( 1) C 6 s(100.00%)p 0.00( 0.00%)
1.0000 0.0002 0.0000 0.0000 0.0002
0.0000 0.0001 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.0000 0.0000
22. (0.00483) RY*( 1) C 1 s( 0.43%)p99.99( 92.53%)d16.27( 7.04%)
0.0000 -0.0135 0.0633 0.0115 0.0229
0.7308 -0.0196 -0.6247 0.0000 0.0000
0.2603 0.0000 0.0000 -0.0411 0.0305
23. (0.00273) RY*( 2) C 1 s( 0.00%)p 1.00( 98.41%)d 0.02( 1.59%)
0.0000 0.0000 -0.0001 0.0000 -0.0256
0.6440 -0.0300 0.7535 0.0000 0.0002
0.0199 0.0000 0.0000 0.1246 0.0000
The following information from the .log file (showing only MO 7-23) shows that all the C-H bonds have the same occupancy and energy, enforcing the idea of aromaticity.
Natural Bond Orbitals (Summary):
Principal Delocalizations
NBO Occupancy Energy (geminal,vicinal,remote)
====================================================================================
Molecular unit 1 (C6H6)
7. BD ( 1) C 2 - H 8 1.98305 -0.51233 113(v),107(v),42(v),22(v),110(g),106(g)
8. BD ( 1) C 3 - C 4 1.98096 -0.68183 110(g),115(g),112(v),119(v),33(v),63(v),114(g),117(g)
62(v),32(v)
9. BD ( 1) C 3 - H 9 1.98305 -0.51236 106(v),115(v),32(v),52(v),110(g),113(g)
10. BD ( 1) C 4 - C 5 1.98098 -0.68200 118(g),113(g),114(v),120(v),73(v),43(v),117(g),119(g)
42(v),72(v)
11. BD ( 2) C 4 - C 5 1.66533 -0.23794 108(v),111(v),45(v),75(v)
12. BD ( 1) C 4 - H 10 1.98305 -0.51236 118(v),110(v),62(v),42(v),115(g),113(g)
13. BD ( 1) C 5 - C 6 1.98096 -0.68186 115(g),107(g),109(v),117(v),53(v),23(v),119(g),120(g)
22(v),52(v)
14. BD ( 1) C 5 - H 11 1.98305 -0.51233 113(v),107(v),52(v),72(v),115(g),118(g)
15. BD ( 1) C 6 - H 12 1.98305 -0.51236 106(v),115(v),22(v),62(v),107(g),118(g)
16. CR ( 1) C 1 1.99911 -10.04057 73(v),33(v),110(v),118(v),120(v),112(v)
17. CR ( 1) C 2 1.99911 -10.04056 43(v),23(v),113(v),107(v),114(v),109(v)
18. CR ( 1) C 3 1.99911 -10.04056 33(v),53(v),106(v),115(v),112(v),117(v)
19. CR ( 1) C 4 1.99911 -10.04057 63(v),43(v),118(v),110(v),119(v),114(v)
20. CR ( 1) C 5 1.99911 -10.04056 53(v),73(v),113(v),107(v),117(v),120(v)
21. CR ( 1) C 6 1.99911 -10.04056 23(v),63(v),115(v),106(v),109(v),119(v)
22. RY*( 1) C 1 0.00483 1.27869
23. RY*( 2) C 1 0.00273 0.71506
Boratabenzene
Boratabenzene is an analogue of benzene, with one C-H group replaced by B-H. In order to remain isoelectronic to benzene, a -1 charge is applied to the molecule.
Optimization
The DFT/B3LYP method was used with 6-31G(d,p) basis set. This file was submitted to D-space: [9]

| X-H length | H-X-C angle | |
|---|---|---|
| X = C |  |

|
| 1.10Å | 120.4° | |
| X = B |  |

|
| 1.22Å | 122.5° |
The C-C and B-C bond lengths are 1.40Å.
The following information from the .log file shows that the parameters converged:
Item Value Threshold Converged?
Maximum Force 0.000246 0.000450 YES
RMS Force 0.000077 0.000300 YES
Maximum Displacement 0.001240 0.001800 YES
RMS Displacement 0.000402 0.001200 YES
Predicted change in Energy=-8.695773D-07
Optimization completed.
-- Stationary point found.
----------------------------
! Optimized Parameters !
! (Angstroms and Degrees) !
-------------------------- --------------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------------------
! R1 R(1,2) 1.4053 -DE/DX = -0.0001 !
! R2 R(1,5) 1.4053 -DE/DX = -0.0001 !
! R3 R(1,6) 1.0917 -DE/DX = -0.0001 !
! R4 R(2,3) 1.3988 -DE/DX = 0.0 !
! R5 R(2,7) 1.0968 -DE/DX = 0.0 !
! R6 R(3,8) 1.0971 -DE/DX = -0.0001 !
! R7 R(3,12) 1.5139 -DE/DX = 0.0001 !
! R8 R(4,5) 1.3989 -DE/DX = 0.0 !
! R9 R(4,10) 1.0971 -DE/DX = -0.0001 !
! R10 R(4,12) 1.5139 -DE/DX = 0.0001 !
! R11 R(5,11) 1.0968 -DE/DX = 0.0 !
! R12 R(9,12) 1.2178 -DE/DX = 0.0002 !
! A1 A(2,1,5) 120.4471 -DE/DX = -0.0001 !
! A2 A(2,1,6) 119.7758 -DE/DX = 0.0001 !
! A3 A(5,1,6) 119.7771 -DE/DX = 0.0001 !
! A4 A(1,2,3) 122.1405 -DE/DX = 0.0001 !
! A5 A(1,2,7) 117.4328 -DE/DX = 0.0 !
! A6 A(3,2,7) 120.4267 -DE/DX = -0.0002 !
! A7 A(2,3,8) 115.9365 -DE/DX = 0.0002 !
! A8 A(2,3,12) 120.0927 -DE/DX = -0.0001 !
! A9 A(8,3,12) 123.9708 -DE/DX = -0.0001 !
! A10 A(5,4,10) 115.9348 -DE/DX = 0.0002 !
! A11 A(5,4,12) 120.0928 -DE/DX = -0.0001 !
! A12 A(10,4,12) 123.9724 -DE/DX = -0.0001 !
! A13 A(1,5,4) 122.1409 -DE/DX = 0.0001 !
! A14 A(1,5,11) 117.4337 -DE/DX = 0.0 !
! A15 A(4,5,11) 120.4254 -DE/DX = -0.0002 !
! A16 A(3,12,4) 115.086 -DE/DX = 0.0 !
! A17 A(3,12,9) 122.4556 -DE/DX = 0.0 !
! A18 A(4,12,9) 122.4584 -DE/DX = 0.0 !
! D1 D(5,1,2,3) -0.0017 -DE/DX = 0.0 !
! D2 D(5,1,2,7) 180.0005 -DE/DX = 0.0 !
! D3 D(6,1,2,3) -180.0013 -DE/DX = 0.0 !
! D4 D(6,1,2,7) 0.0009 -DE/DX = 0.0 !
! D5 D(2,1,5,4) 0.0003 -DE/DX = 0.0 !
! D6 D(2,1,5,11) 180.0011 -DE/DX = 0.0 !
! D7 D(6,1,5,4) 179.9998 -DE/DX = 0.0 !
! D8 D(6,1,5,11) 0.0007 -DE/DX = 0.0 !
! D9 D(1,2,3,8) 180.0018 -DE/DX = 0.0 !
! D10 D(1,2,3,12) 0.002 -DE/DX = 0.0 !
! D11 D(7,2,3,8) -0.0005 -DE/DX = 0.0 !
! D12 D(7,2,3,12) -180.0004 -DE/DX = 0.0 !
! D13 D(2,3,12,4) -0.0008 -DE/DX = 0.0 !
! D14 D(2,3,12,9) -180.0007 -DE/DX = 0.0 !
! D15 D(8,3,12,4) -180.0006 -DE/DX = 0.0 !
! D16 D(8,3,12,9) -0.0006 -DE/DX = 0.0 !
! D17 D(10,4,5,1) -179.9997 -DE/DX = 0.0 !
! D18 D(10,4,5,11) -0.0005 -DE/DX = 0.0 !
! D19 D(12,4,5,1) 0.0009 -DE/DX = 0.0 !
! D20 D(12,4,5,11) 180.0001 -DE/DX = 0.0 !
! D21 D(5,4,12,3) -0.0006 -DE/DX = 0.0 !
! D22 D(5,4,12,9) -180.0006 -DE/DX = 0.0 !
! D23 D(10,4,12,3) -180.0 -DE/DX = 0.0 !
! D24 D(10,4,12,9) 0.0 -DE/DX = 0.0 !
--------------------------------------------------------------------------------
GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
The data above shows that the optimized boratabenzene molecule has C-H bond lengths of 1.10Å, H-C-C bond angles of 120.4°, B-H bond lengths of 1.22Å, H-B-C bond angles of 122.5°, and C-C and B-C bond lengths of 1.40Å. The symmetry group is C1 and has a 2.8445 Debye dipole moment as it is not highly symmetric. The job took 4 min 4.7 seconds to run. The frequency analysis is run next to determine if the minima was found.
Frequency Analysis
The same method and basis set were used, with job = frequency. This frequency analysis gave the following files which were submitted to D-space: [10].

| X-H length | H-X-C angle | |
|---|---|---|
| X = C |  |

|
| 1.10Å | 120.4° | |
| X = B |  |

|
| 1.22Å | 122.5° |
The frequency calculation was successful, and the following data shows that all the parameters converged:
Item Value Threshold Converged?
Maximum Force 0.000439 0.000450 YES
RMS Force 0.000162 0.000300 YES
Maximum Displacement 0.001088 0.001800 YES
RMS Displacement 0.000490 0.001200 YES
Predicted change in Energy=-9.399944D-07
Optimization completed.
-- Stationary point found.
GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
Low frequencies --- -14.5246 -0.0010 -0.0009 -0.0004 15.8790 18.0052 Low frequencies --- 371.2523 404.2379 565.1702
Vibrational analysis
| Mode | Frequency (cm-1) | Intensity | Mode | Frequency (cm-1) | Intensity | Mode | Frequency (cm-1) | Intensity |
|---|---|---|---|---|---|---|---|---|
| 1 | 371 | 2 | 11 | 917 | 1 | 21 | 1449 | 9 |
| 2 | 404 | 0 | 12 | 951 | 0 | 22 | 1463 | 14 |
| 3 | 565 | 0 | 13 | 951 | 0 | 23 | 1565 | 7 |
| 4 | 569 | 0 | 14 | 960 | 2 | 24 | 1592 | 40 |
| 5 | 608 | 11 | 15 | 1012 | 4 | 25 | 2451 | 367 |
| 6 | 711 | 3 | 16 | 1085 | 3 | 26 | 3028 | 109 |
| 7 | 757 | 8 | 17 | 1175 | 1 | 27 | 3030 | 2 |
| 8 | 814 | 0 | 18 | 1180 | 1 | 28 | 3060 | 378 |
| 9 | 873 | 28 | 19 | 1227 | 1 | 29 | 3061 | 8 |
| 10 | 906 | 0 | 20 | 1333 | 31 | 30 | 3116 | 113 |
This gives the following IR spectrum:

There are many vibrational modes that are either inactive or have an intensity so low that it does not show up on the spectrum. Modes 5, 9, 20-26, 28, and 30 are the strongest modes with the highest intensities. The rest of the peaks are from the modes that give low intensities.
MO analysis
The link to D-space can be found here: [11].
There is no degeneracy since the symmetry is reduced due to the B-H in the molecule. The energy of the HOMO is 0.01095 a.u. and the energy of the LUMO is 0.21372 a.u..
Charge distribution
An NBO analysis was run on the .log file of the optimized boratazene molecule. The color range was from -0.250 to +0.250.


The relative charges were -0.224 for the H attached to B, -0.041 for the ortho-Hs, -0.026 for the meta-Hs, -0.027 for the para-H, -0.070 for B, -0.108 for all C except for para-C, and -0.113 for the para-C.
The following part of the .log file shows the natural charge information:
Summary of Natural Population Analysis:
Natural Population
Natural -----------------------------------------------
Atom No Charge Core Valence Rydberg Total
-----------------------------------------------------------------------
C 1 -0.33983 1.99907 4.32692 0.01384 6.33983
C 2 -0.25045 1.99910 4.23723 0.01412 6.25045
C 3 -0.58783 1.99901 4.57703 0.01178 6.58783
C 4 -0.58785 1.99901 4.57705 0.01178 6.58785
C 5 -0.25044 1.99910 4.23722 0.01412 6.25044
H 6 0.18563 0.00000 0.81238 0.00200 0.81437
H 7 0.17905 0.00000 0.81833 0.00262 0.82095
H 8 0.18376 0.00000 0.81406 0.00218 0.81624
H 9 -0.09633 0.00000 1.09579 0.00054 1.09633
H 10 0.18376 0.00000 0.81406 0.00218 0.81624
H 11 0.17905 0.00000 0.81833 0.00262 0.82095
B 12 0.20148 1.99906 2.78787 0.01159 4.79852
=======================================================================
* Total * -1.00000 11.99436 29.91625 0.08939 42.00000
The following information from the .log file (up to the LUMO) gives precise details on electron density.
(Occupancy) Bond orbital/ Coefficients/ Hybrids
---------------------------------------------------------------------------------
1. (1.97970) BD ( 1) C 1 - C 2
( 50.04%) 0.7074* C 1 s( 35.88%)p 1.79( 64.09%)d 0.00( 0.04%)
-0.0001 0.5989 -0.0072 0.0010 0.7062
0.0327 -0.3753 0.0141 0.0000 0.0000
-0.0137 0.0000 0.0000 0.0078 -0.0107
( 49.96%) 0.7068* C 2 s( 35.50%)p 1.82( 64.46%)d 0.00( 0.04%)
-0.0001 0.5958 -0.0075 0.0006 -0.6874
-0.0034 0.4134 0.0325 0.0000 0.0000
-0.0146 0.0000 0.0000 0.0081 -0.0107
2. (1.97970) BD ( 1) C 1 - C 5
( 50.04%) 0.7074* C 1 s( 35.88%)p 1.79( 64.09%)d 0.00( 0.04%)
-0.0001 0.5989 -0.0072 0.0010 -0.7062
-0.0327 -0.3753 0.0141 0.0000 0.0000
0.0137 0.0000 0.0000 0.0078 -0.0107
( 49.96%) 0.7068* C 5 s( 35.50%)p 1.82( 64.46%)d 0.00( 0.04%)
-0.0001 0.5958 -0.0075 0.0006 0.6875
0.0034 0.4134 0.0325 0.0000 0.0000
0.0146 0.0000 0.0000 0.0081 -0.0107
3. (1.98507) BD ( 1) C 1 - H 6
( 59.44%) 0.7710* C 1 s( 28.22%)p 2.54( 71.74%)d 0.00( 0.04%)
-0.0004 0.5311 0.0116 -0.0020 0.0000
0.0000 0.8469 -0.0076 0.0000 0.0000
0.0000 0.0000 0.0000 -0.0178 -0.0110
( 40.56%) 0.6369* H 6 s( 99.95%)p 0.00( 0.05%)
0.9998 0.0011 0.0000 -0.0217 0.0000
4. (1.98271) BD ( 1) C 2 - C 3
( 50.77%) 0.7125* C 2 s( 37.60%)p 1.66( 62.37%)d 0.00( 0.03%)
-0.0001 0.6131 -0.0079 0.0007 0.0574
0.0311 -0.7868 -0.0165 0.0000 0.0000
-0.0020 0.0000 0.0000 -0.0150 -0.0098
( 49.23%) 0.7017* C 3 s( 32.50%)p 2.08( 67.46%)d 0.00( 0.05%)
0.0000 0.5697 -0.0200 0.0010 -0.0027
0.0269 0.8201 0.0353 0.0000 0.0000
0.0006 0.0000 0.0000 -0.0173 -0.0123
5. (1.76869) BD ( 2) C 2 - C 3
( 48.13%) 0.6938* C 2 s( 0.00%)p 1.00( 99.97%)d 0.00( 0.03%)
0.0000 0.0000 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.9996 -0.0213
0.0000 -0.0031 -0.0171 0.0000 0.0000
( 51.87%) 0.7202* C 3 s( 0.00%)p 1.00( 99.97%)d 0.00( 0.03%)
0.0000 0.0000 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.9998 -0.0054
0.0000 -0.0016 0.0185 0.0000 0.0000
6. (1.98569) BD ( 1) C 2 - H 7
( 59.32%) 0.7702* C 2 s( 26.87%)p 2.72( 73.08%)d 0.00( 0.05%)
-0.0003 0.5182 0.0133 -0.0012 0.7228
-0.0089 0.4562 -0.0100 0.0000 0.0000
0.0177 0.0000 0.0000 0.0069 -0.0111
( 40.68%) 0.6378* H 7 s( 99.95%)p 0.00( 0.05%)
0.9998 0.0026 -0.0187 -0.0116 0.0000
7. (1.98420) BD ( 1) C 3 - H 8
( 59.41%) 0.7708* C 3 s( 25.38%)p 2.94( 74.57%)d 0.00( 0.05%)
0.0003 -0.5038 0.0050 0.0025 -0.7907
0.0003 0.3470 0.0088 0.0000 0.0000
0.0111 0.0000 0.0000 -0.0150 0.0119
( 40.59%) 0.6371* H 8 s( 99.95%)p 0.00( 0.05%)
-0.9998 -0.0005 0.0192 -0.0100 0.0000
8. (1.96996) BD ( 1) C 3 - B 12
( 66.70%) 0.8167* C 3 s( 42.03%)p 1.38( 57.96%)d 0.00( 0.01%)
0.0000 -0.6481 -0.0158 -0.0012 0.6115
-0.0293 0.4524 0.0090 0.0000 0.0000
-0.0059 0.0000 0.0000 -0.0041 0.0057
( 33.30%) 0.5771* B 12 s( 33.39%)p 1.99( 66.54%)d 0.00( 0.08%)
0.0000 -0.5778 0.0059 -0.0049 -0.7057
-0.0393 -0.4071 0.0096 0.0000 0.0000
-0.0230 0.0000 0.0000 -0.0082 0.0133
9. (1.98271) BD ( 1) C 4 - C 5
( 49.23%) 0.7017* C 4 s( 32.50%)p 2.08( 67.46%)d 0.00( 0.05%)
0.0000 0.5697 -0.0200 0.0010 0.0027
-0.0269 0.8201 0.0353 0.0000 0.0000
-0.0006 0.0000 0.0000 -0.0173 -0.0123
( 50.77%) 0.7125* C 5 s( 37.60%)p 1.66( 62.37%)d 0.00( 0.03%)
-0.0001 0.6131 -0.0079 0.0007 -0.0574
-0.0311 -0.7868 -0.0164 0.0000 0.0000
0.0020 0.0000 0.0000 -0.0150 -0.0098
10. (1.76867) BD ( 2) C 4 - C 5
( 51.87%) 0.7202* C 4 s( 0.00%)p 1.00( 99.97%)d 0.00( 0.03%)
0.0000 0.0000 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.9998 -0.0054
0.0000 0.0016 0.0185 0.0000 0.0000
( 48.13%) 0.6938* C 5 s( 0.00%)p 1.00( 99.97%)d 0.00( 0.03%)
0.0000 0.0000 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.9996 -0.0213
0.0000 0.0031 -0.0171 0.0000 0.0000
11. (1.98420) BD ( 1) C 4 - H 10
( 59.41%) 0.7708* C 4 s( 25.38%)p 2.94( 74.57%)d 0.00( 0.05%)
0.0003 -0.5038 0.0050 0.0025 0.7908
-0.0003 0.3469 0.0088 0.0000 0.0000
-0.0111 0.0000 0.0000 -0.0150 0.0119
( 40.59%) 0.6371* H 10 s( 99.95%)p 0.00( 0.05%)
-0.9998 -0.0005 -0.0192 -0.0100 0.0000
12. (1.96997) BD ( 1) C 4 - B 12
( 66.70%) 0.8167* C 4 s( 42.03%)p 1.38( 57.96%)d 0.00( 0.01%)
0.0000 0.6481 0.0158 0.0012 0.6115
-0.0293 -0.4525 -0.0090 0.0000 0.0000
-0.0059 0.0000 0.0000 0.0041 -0.0057
( 33.30%) 0.5771* B 12 s( 33.39%)p 1.99( 66.53%)d 0.00( 0.08%)
0.0000 0.5778 -0.0059 0.0049 -0.7056
-0.0393 0.4072 -0.0096 0.0000 0.0000
-0.0230 0.0000 0.0000 0.0082 -0.0133
13. (1.98569) BD ( 1) C 5 - H 11
( 59.32%) 0.7702* C 5 s( 26.87%)p 2.72( 73.08%)d 0.00( 0.05%)
0.0003 -0.5182 -0.0133 0.0012 0.7228
-0.0089 -0.4563 0.0101 0.0000 0.0000
0.0177 0.0000 0.0000 -0.0069 0.0111
( 40.68%) 0.6378* H 11 s( 99.95%)p 0.00( 0.05%)
-0.9998 -0.0026 -0.0187 0.0116 0.0000
14. (1.98605) BD ( 1) H 9 - B 12
( 55.08%) 0.7422* H 9 s( 99.97%)p 0.00( 0.03%)
0.9998 0.0001 0.0000 0.0181 0.0000
( 44.92%) 0.6702* B 12 s( 33.19%)p 2.01( 66.76%)d 0.00( 0.06%)
-0.0005 0.5760 0.0069 -0.0061 0.0000
0.0000 -0.8170 0.0015 0.0000 0.0000
0.0000 0.0000 0.0000 -0.0213 -0.0105
15. (1.99907) CR ( 1) C 1 s(100.00%)p 0.00( 0.00%)
1.0000 0.0003 0.0000 0.0000 0.0000
0.0000 0.0002 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.0000 0.0000
16. (1.99910) CR ( 1) C 2 s(100.00%)p 0.00( 0.00%)
1.0000 0.0003 0.0000 0.0000 0.0002
0.0000 0.0001 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.0000 0.0000
17. (1.99902) CR ( 1) C 3 s(100.00%)p 0.00( 0.00%)
1.0000 0.0001 0.0000 0.0000 0.0002
0.0000 -0.0002 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.0000 0.0000
18. (1.99902) CR ( 1) C 4 s(100.00%)p 0.00( 0.00%)
1.0000 0.0001 0.0000 0.0000 -0.0002
0.0000 -0.0002 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.0000 0.0000
19. (1.99910) CR ( 1) C 5 s(100.00%)p 0.00( 0.00%)
1.0000 0.0003 0.0000 0.0000 -0.0002
0.0000 0.0001 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.0000 0.0000
20. (1.99907) CR ( 1) B 12 s(100.00%)p 0.00( 0.00%)
1.0000 0.0003 0.0000 0.0000 0.0000
0.0000 -0.0004 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.0000 0.0000
21. (1.14687) LP ( 1) C 1 s( 0.00%)p 1.00( 99.99%)d 0.00( 0.01%)
0.0000 0.0000 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.9996 -0.0282
0.0000 0.0000 -0.0088 0.0000 0.0000
22. (0.57259) LP*( 1) B 12 s( 0.00%)p 1.00( 99.96%)d 0.00( 0.04%)
0.0000 0.0000 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.9984 -0.0535
0.0000 0.0000 0.0199 0.0000 0.0000
The following information from the .log file (up to the LUMO).
Natural Bond Orbitals (Summary):
Principal Delocalizations
NBO Occupancy Energy (geminal,vicinal,remote)
====================================================================================
Molecular unit 1 (C5H6B)
1. BD ( 1) C 1 - C 2 1.97970 -0.46973 110(g),108(g),119(v),113(v),64(v),43(v),109(g),112(g)
2. BD ( 1) C 1 - C 5 1.97970 -0.46975 115(g),107(g),112(v),117(v),34(v),53(v),109(g),119(g)
3. BD ( 1) C 1 - H 6 1.98507 -0.31742 115(v),110(v),33(v),63(v),108(g),107(g)
4. BD ( 1) C 2 - C 3 1.98271 -0.46501 114(g),107(g),109(v),112(g),24(v),120(v),113(g),98(v),23(v)
5. BD ( 2) C 2 - C 3 1.76869 -0.02907 21(v),22(v),25(v),100(v),111(g)
6. BD ( 1) C 2 - H 7 1.98569 -0.31409 108(v),114(v),23(v),43(v),110(g)
7. BD ( 1) C 3 - H 8 1.98420 -0.28844 107(v),114(g),33(v),118(v),97(v),110(g)
8. BD ( 1) C 3 - B 12 1.96996 -0.31769 110(g),112(v),117(v),113(g),34(v),33(v),81(v),54(v),118(g)
9. BD ( 1) C 4 - C 5 1.98271 -0.46499 118(g),108(g),109(v),119(g),24(v),120(v),117(g),98(v),23(v)
10. BD ( 2) C 4 - C 5 1.76867 -0.02907 21(v),22(v),25(v),100(v),116(g)
11. BD ( 1) C 4 - H 10 1.98420 -0.28843 108(v),118(g),63(v),114(v),97(v),115(g)
12. BD ( 1) C 4 - B 12 1.96997 -0.31770 115(g),119(v),113(v),117(g),64(v),63(v),89(v),44(v),114(g)
13. BD ( 1) C 5 - H 11 1.98569 -0.31409 107(v),118(v),23(v),53(v),115(g)
14. BD ( 1) H 9 - B 12 1.98605 -0.17273 115(v),110(v),43(v),53(v)
15. CR ( 1) C 1 1.99907 -9.82825 34(v),64(v),110(v),115(v),112(v),119(v),37(v),67(v),63(v),33(v)
16. CR ( 1) C 2 1.99910 -9.83476 44(v),24(v),114(v),109(v),108(v),113(v)
17. CR ( 1) C 3 1.99902 -9.79410 34(v),98(v),114(g),107(v),112(v),97(v)
18. CR ( 1) C 4 1.99902 -9.79410 64(v),98(v),118(g),108(v),119(v),97(v)
19. CR ( 1) C 5 1.99910 -9.83476 54(v),24(v),118(v),109(v),107(v),117(v)
20. CR ( 1) B 12 1.99907 -6.36939 117(v),113(v),115(v),110(v),53(v),43(v)
21. LP ( 1) C 1 1.14687 0.09691 116(v),111(v),25(g),66(v),36(v),35(v),65(v)
22. LP*( 1) B 12 0.57259 0.22265 116(v),111(v),100(g),57(v),47(v)
23. RY*( 1) C 1 0.00435 1.45199
Pyridinium
Pyridinium is an analogue of benzene, with one C-H group replaced by N-H. In order to remain isoelectronic to benzene, a +1 charge is applied to the molecule.
Optimization
The method used was DFT/B3LYP with 6-31G(d,p) basis set. This file was submitted to D-space: [12]

| X-H length | H-X-C angle | |
|---|---|---|
| X = C |  |

|
| 1.02Å | 118.3° | |
| X = N |  |

|
| 1.08Å | 121.5° |
The following information from the .log file shows that the parameters converged:
Item Value Threshold Converged?
Maximum Force 0.000065 0.000450 YES
RMS Force 0.000023 0.000300 YES
Maximum Displacement 0.000826 0.001800 YES
RMS Displacement 0.000176 0.001200 YES
Predicted change in Energy=-6.972574D-08
Optimization completed.
-- Stationary point found.
----------------------------
! Optimized Parameters !
! (Angstroms and Degrees) !
-------------------------- --------------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------------------
! R1 R(1,2) 1.3988 -DE/DX = 0.0 !
! R2 R(1,5) 1.3838 -DE/DX = 0.0 !
! R3 R(1,6) 1.0835 -DE/DX = 0.0 !
! R4 R(2,3) 1.3988 -DE/DX = 0.0 !
! R5 R(2,7) 1.0852 -DE/DX = 0.0 !
! R6 R(3,4) 1.3839 -DE/DX = 0.0 !
! R7 R(3,8) 1.0835 -DE/DX = 0.0 !
! R8 R(4,9) 1.0832 -DE/DX = 0.0 !
! R9 R(4,12) 1.3523 -DE/DX = 0.0001 !
! R10 R(5,11) 1.0832 -DE/DX = 0.0 !
! R11 R(5,12) 1.3524 -DE/DX = 0.0 !
! R12 R(10,12) 1.0169 -DE/DX = 0.0 !
! A1 A(2,1,5) 119.0827 -DE/DX = 0.0 !
! A2 A(2,1,6) 121.496 -DE/DX = -0.0001 !
! A3 A(5,1,6) 119.4213 -DE/DX = 0.0 !
! A4 A(1,2,3) 120.0549 -DE/DX = 0.0 !
! A5 A(1,2,7) 119.9711 -DE/DX = 0.0 !
! A6 A(3,2,7) 119.974 -DE/DX = 0.0 !
! A7 A(2,3,4) 119.082 -DE/DX = 0.0 !
! A8 A(2,3,8) 121.4988 -DE/DX = -0.0001 !
! A9 A(4,3,8) 119.4192 -DE/DX = 0.0001 !
! A10 A(3,4,9) 123.9297 -DE/DX = 0.0 !
! A11 A(3,4,12) 119.2363 -DE/DX = 0.0 !
! A12 A(9,4,12) 116.834 -DE/DX = 0.0 !
! A13 A(1,5,11) 123.9327 -DE/DX = 0.0 !
! A14 A(1,5,12) 119.2354 -DE/DX = 0.0 !
! A15 A(11,5,12) 116.8319 -DE/DX = 0.0 !
! A16 A(4,12,5) 123.3088 -DE/DX = 0.0 !
! A17 A(4,12,10) 118.3463 -DE/DX = 0.0 !
! A18 A(5,12,10) 118.345 -DE/DX = 0.0 !
! D1 D(5,1,2,3) 0.0006 -DE/DX = 0.0 !
! D2 D(5,1,2,7) -180.0023 -DE/DX = 0.0 !
! D3 D(6,1,2,3) 180.0015 -DE/DX = 0.0 !
! D4 D(6,1,2,7) -0.0014 -DE/DX = 0.0 !
! D5 D(2,1,5,11) -180.0005 -DE/DX = 0.0 !
! D6 D(2,1,5,12) 0.0021 -DE/DX = 0.0 !
! D7 D(6,1,5,11) -0.0014 -DE/DX = 0.0 !
! D8 D(6,1,5,12) 180.0012 -DE/DX = 0.0 !
! D9 D(1,2,3,4) -0.0024 -DE/DX = 0.0 !
! D10 D(1,2,3,8) -180.0018 -DE/DX = 0.0 !
! D11 D(7,2,3,4) 180.0005 -DE/DX = 0.0 !
! D12 D(7,2,3,8) 0.0012 -DE/DX = 0.0 !
! D13 D(2,3,4,9) 180.0002 -DE/DX = 0.0 !
! D14 D(2,3,4,12) 0.0015 -DE/DX = 0.0 !
! D15 D(8,3,4,9) -0.0004 -DE/DX = 0.0 !
! D16 D(8,3,4,12) 180.0008 -DE/DX = 0.0 !
! D17 D(3,4,12,5) 0.0014 -DE/DX = 0.0 !
! D18 D(3,4,12,10) -180.001 -DE/DX = 0.0 !
! D19 D(9,4,12,5) 180.0025 -DE/DX = 0.0 !
! D20 D(9,4,12,10) 0.0001 -DE/DX = 0.0 !
! D21 D(1,5,12,4) -0.0032 -DE/DX = 0.0 !
! D22 D(1,5,12,10) -180.0008 -DE/DX = 0.0 !
! D23 D(11,5,12,4) -180.0007 -DE/DX = 0.0 !
! D24 D(11,5,12,10) 0.0016 -DE/DX = 0.0 !
--------------------------------------------------------------------------------
GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
The data above shows that the optimized pyridinium molecule has C-H bond lengths of 1.02Å, H-C-C bond angles of 118.3°, N-H bond lengths of 1.08Å, H-B-C bond angles of 121.5°, and C-C and B-C bond lengths of 1.40Å. The symmetry group is C1 and has a 1.87 Debye dipole moment as it is not highly symmetric. The job took 3 min 59.7 seconds to run. The frequency analysis is run next to determine if the minima was found.
Frequency Analysis
The same method and basis set were used, with job = frequency. This frequency analysis was submitted to D-space: [13].

| X-H length | H-X-C angle | |
|---|---|---|
| X = C |  |

|
| 1.08Å | 121.5° | |
| X = N |  |

|
| 1.02Å | 118.3° |
The frequency calculation was successful, and the following data shows that all the parameters converged:
Item Value Threshold Converged?
Maximum Force 0.000156 0.000450 YES
RMS Force 0.000039 0.000300 YES
Maximum Displacement 0.000778 0.001800 YES
RMS Displacement 0.000229 0.001200 YES
Predicted change in Energy=-7.500721D-08
Optimization completed.
-- Stationary point found.
GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
Low frequencies --- -7.2115 -0.0005 -0.0004 0.0010 17.3444 18.5499 Low frequencies --- 392.4572 404.0614 620.4717
Vibrational analysis
| Mode | Frequency (cm-1) | Intensity | Mode | Frequency (cm-1) | Intensity | Mode | Frequency (cm-1) | Intensity |
|---|---|---|---|---|---|---|---|---|
| 1 | 392 | 1 | 11 | 1023 | 4 | 21 | 1524 | 21 |
| 2 | 404 | 0 | 12 | 1048 | 0 | 22 | 1580 | 48 |
| 3 | 620 | 0 | 13 | 1052 | 0 | 23 | 1657 | 32 |
| 4 | 645 | 0 | 14 | 1082 | 3 | 24 | 1677 | 34 |
| 5 | 677 | 89 | 15 | 1087 | 4 | 25 | 3225 | 0 |
| 6 | 748 | 83 | 16 | 1200 | 3 | 26 | 3241 | 1 |
| 7 | 855 | 11 | 17 | 1229 | 2 | 27 | 3243 | 11 |
| 8 | 882 | 0 | 18 | 1230 | 3 | 28 | 3253 | 21 |
| 9 | 992 | 2 | 19 | 1374 | 12 | 29 | 3254 | 0 |
| 10 | 1005 | 0 | 20 | 1416 | 3 | 30 | 3569 | 158 |
This gives the following IR spectrum:

Again there are quite a few modes that are inactive or have very low intensity. The highest intensity modes are: 5-7, 19, 21-24, 27-28, and 30.
MO analysis
The same method and basis set were used, with job = energy. The link to D-space can be found here: [[14]]
There is no degeneracy since the symmetry is reduced due to the N-H in the molecule. The energy of the HOMO is -0.00495 a.u. and the energy of the LUMO is 0.01202 a.u..
Charge distribution
An NBO analysis was run on the .log file of the optimized pyridinium molecule. The color range was from -0.500 to +0.500.


The relative charges were 0.483 for the H attached to N, 0.285 for the ortho-Hs, 0.297 for the meta-Hs, 0.292 for the para-H, -0.476 for N, 0.071 for ortho-Cs, -0.241 for meta-Cs, and -0.122 for the para-C.
The following part of the .log file shows the natural charge information:
Summary of Natural Population Analysis:
Natural Population
Natural -----------------------------------------------
Atom No Charge Core Valence Rydberg Total
-----------------------------------------------------------------------
C 1 -0.24103 1.99912 4.22860 0.01331 6.24103
C 2 -0.12241 1.99913 4.10941 0.01386 6.12241
C 3 -0.24104 1.99912 4.22860 0.01331 6.24104
C 4 0.07100 1.99918 3.91066 0.01916 5.92900
C 5 0.07098 1.99918 3.91068 0.01916 5.92902
H 6 0.29718 0.00000 0.70179 0.00103 0.70282
H 7 0.29170 0.00000 0.70718 0.00113 0.70830
H 8 0.29718 0.00000 0.70179 0.00103 0.70282
H 9 0.28493 0.00000 0.71397 0.00110 0.71507
H 10 0.48279 0.00000 0.51475 0.00246 0.51721
H 11 0.28493 0.00000 0.71397 0.00110 0.71507
N 12 -0.47622 1.99937 5.46756 0.00929 7.47622
=======================================================================
* Total * 1.00000 11.99510 29.90895 0.09595 42.00000
The following information from the .log file (up to the LUMO) gives precise details on electron density.
(Occupancy) Bond orbital/ Coefficients/ Hybrids
---------------------------------------------------------------------------------
1. (1.98249) BD ( 1) C 1 - C 2
( 50.26%) 0.7089* C 1 s( 34.73%)p 1.88( 65.23%)d 0.00( 0.04%)
0.0000 0.5893 -0.0066 0.0009 -0.4185
-0.0371 -0.6897 0.0068 0.0000 0.0000
0.0122 0.0000 0.0000 -0.0118 -0.0115
( 49.74%) 0.7053* C 2 s( 34.45%)p 1.90( 65.51%)d 0.00( 0.04%)
0.0000 0.5869 -0.0086 0.0005 0.3935
-0.0234 0.7063 0.0290 0.0000 0.0000
0.0169 0.0000 0.0000 -0.0060 -0.0113
2. (1.98297) BD ( 1) C 1 - C 5
( 49.58%) 0.7042* C 1 s( 33.47%)p 1.99( 66.48%)d 0.00( 0.05%)
0.0000 0.5784 -0.0119 -0.0002 0.8145
0.0194 -0.0009 0.0320 0.0000 0.0000
-0.0047 0.0000 0.0000 0.0179 -0.0119
( 50.42%) 0.7100* C 5 s( 38.49%)p 1.60( 61.47%)d 0.00( 0.04%)
-0.0001 0.6204 -0.0023 0.0030 -0.7832
-0.0046 0.0142 0.0331 0.0000 0.0000
0.0053 0.0000 0.0000 0.0168 -0.0095
3. (1.61445) BD ( 2) C 1 - C 5
( 52.23%) 0.7227* C 1 s( 0.00%)p 1.00( 99.94%)d 0.00( 0.06%)
0.0000 0.0000 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.9997 -0.0068
0.0000 0.0191 -0.0159 0.0000 0.0000
( 47.77%) 0.6912* C 5 s( 0.00%)p 1.00( 99.94%)d 0.00( 0.06%)
0.0000 0.0000 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.9995 -0.0175
0.0000 -0.0196 -0.0153 0.0000 0.0000
4. (1.97822) BD ( 1) C 1 - H 6
( 64.83%) 0.8052* C 1 s( 31.78%)p 2.15( 68.19%)d 0.00( 0.03%)
-0.0003 0.5636 0.0138 -0.0005 -0.3988
0.0072 0.7229 -0.0181 0.0000 0.0000
-0.0109 0.0000 0.0000 -0.0085 -0.0099
( 35.17%) 0.5930* H 6 s( 99.94%)p 0.00( 0.06%)
0.9997 0.0016 0.0116 -0.0208 0.0000
5. (1.98249) BD ( 1) C 2 - C 3
( 49.74%) 0.7053* C 2 s( 34.45%)p 1.90( 65.51%)d 0.00( 0.04%)
0.0000 0.5869 -0.0086 0.0005 0.3934
-0.0234 -0.7064 -0.0290 0.0000 0.0000
-0.0169 0.0000 0.0000 -0.0060 -0.0113
( 50.26%) 0.7089* C 3 s( 34.73%)p 1.88( 65.23%)d 0.00( 0.04%)
0.0000 0.5893 -0.0066 0.0009 -0.4184
-0.0371 0.6898 -0.0068 0.0000 0.0000
-0.0122 0.0000 0.0000 -0.0118 -0.0115
6. (1.54879) BD ( 2) C 2 - C 3
( 45.73%) 0.6762* C 2 s( 0.00%)p 1.00( 99.93%)d 0.00( 0.07%)
0.0000 0.0000 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.9997 -0.0036
0.0000 0.0241 -0.0101 0.0000 0.0000
( 54.27%) 0.7367* C 3 s( 0.00%)p 1.00( 99.94%)d 0.00( 0.06%)
0.0000 0.0000 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.9997 -0.0080
0.0000 0.0086 0.0228 0.0000 0.0000
7. (1.98141) BD ( 1) C 2 - H 7
( 64.64%) 0.8040* C 2 s( 31.07%)p 2.22( 68.89%)d 0.00( 0.03%)
0.0003 -0.5573 -0.0131 0.0007 0.8298
-0.0198 -0.0001 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 -0.0153 0.0101
( 35.36%) 0.5947* H 7 s( 99.94%)p 0.00( 0.06%)
-0.9997 -0.0018 -0.0242 0.0000 0.0000
8. (1.98297) BD ( 1) C 3 - C 4
( 49.58%) 0.7042* C 3 s( 33.47%)p 1.99( 66.48%)d 0.00( 0.05%)
0.0000 0.5784 -0.0119 -0.0002 0.8145
0.0194 0.0007 -0.0320 0.0000 0.0000
0.0047 0.0000 0.0000 0.0179 -0.0119
( 50.42%) 0.7100* C 4 s( 38.49%)p 1.60( 61.47%)d 0.00( 0.04%)
-0.0001 0.6204 -0.0023 0.0030 -0.7832
-0.0046 -0.0140 -0.0331 0.0000 0.0000
-0.0053 0.0000 0.0000 0.0168 -0.0095
9. (1.97822) BD ( 1) C 3 - H 8
( 64.83%) 0.8052* C 3 s( 31.78%)p 2.15( 68.19%)d 0.00( 0.03%)
0.0003 -0.5636 -0.0138 0.0005 0.3989
-0.0072 0.7228 -0.0181 0.0000 0.0000
-0.0109 0.0000 0.0000 0.0085 0.0099
( 35.17%) 0.5930* H 8 s( 99.94%)p 0.00( 0.06%)
-0.9997 -0.0016 -0.0116 -0.0208 0.0000
10. (1.98154) BD ( 1) C 4 - H 9
( 64.26%) 0.8016* C 4 s( 33.44%)p 1.99( 66.52%)d 0.00( 0.04%)
0.0004 -0.5780 -0.0180 0.0017 -0.4693
0.0193 0.6666 -0.0183 0.0000 0.0000
0.0164 0.0000 0.0000 0.0019 0.0092
( 35.74%) 0.5978* H 9 s( 99.94%)p 0.00( 0.06%)
-0.9997 -0.0018 0.0128 -0.0209 0.0000
11. (1.98861) BD ( 1) C 4 - N 12
( 36.68%) 0.6057* C 4 s( 28.13%)p 2.55( 71.74%)d 0.00( 0.13%)
-0.0001 0.5293 -0.0335 -0.0013 0.4043
0.0563 0.7416 0.0277 0.0000 0.0000
0.0252 0.0000 0.0000 -0.0185 -0.0179
( 63.32%) 0.7957* N 12 s( 36.56%)p 1.73( 63.41%)d 0.00( 0.03%)
-0.0001 0.6047 -0.0037 0.0006 -0.3660
0.0187 -0.7069 -0.0132 0.0000 0.0000
0.0107 0.0000 0.0000 -0.0059 -0.0115
12. (1.82447) BD ( 2) C 4 - N 12
( 28.55%) 0.5343* C 4 s( 0.00%)p 1.00( 99.83%)d 0.00( 0.17%)
0.0000 0.0000 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.9991 0.0132
0.0000 0.0102 0.0394 0.0000 0.0000
( 71.45%) 0.8453* N 12 s( 0.00%)p 1.00( 99.98%)d 0.00( 0.02%)
0.0000 0.0000 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.9999 0.0036
0.0000 -0.0128 -0.0077 0.0000 0.0000
13. (1.98154) BD ( 1) C 5 - H 11
( 64.26%) 0.8016* C 5 s( 33.44%)p 1.99( 66.52%)d 0.00( 0.04%)
-0.0004 0.5780 0.0180 -0.0017 0.4694
-0.0193 0.6664 -0.0183 0.0000 0.0000
0.0164 0.0000 0.0000 -0.0019 -0.0092
( 35.74%) 0.5978* H 11 s( 99.94%)p 0.00( 0.06%)
0.9997 0.0018 -0.0128 -0.0209 0.0000
14. (1.98861) BD ( 1) C 5 - N 12
( 36.68%) 0.6057* C 5 s( 28.13%)p 2.55( 71.74%)d 0.00( 0.13%)
-0.0001 0.5293 -0.0335 -0.0013 0.4042
0.0563 -0.7417 -0.0277 0.0000 0.0000
-0.0252 0.0000 0.0000 -0.0185 -0.0179
( 63.32%) 0.7957* N 12 s( 36.56%)p 1.73( 63.41%)d 0.00( 0.03%)
-0.0001 0.6046 -0.0037 0.0006 -0.3658
0.0187 0.7069 0.0132 0.0000 0.0000
-0.0107 0.0000 0.0000 -0.0059 -0.0115
15. (1.98630) BD ( 1) H 10 - N 12
( 25.41%) 0.5041* H 10 s( 99.88%)p 0.00( 0.12%)
0.9994 -0.0064 -0.0342 0.0000 0.0000
( 74.59%) 0.8637* N 12 s( 26.82%)p 2.73( 73.16%)d 0.00( 0.02%)
-0.0002 0.5178 0.0066 -0.0013 0.8553
-0.0091 -0.0001 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.0115 -0.0106
16. (1.99913) CR ( 1) C 1 s(100.00%)p 0.00( 0.00%)
1.0000 0.0002 0.0000 0.0000 -0.0001
0.0000 0.0002 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.0000 0.0000
17. (1.99914) CR ( 1) C 2 s(100.00%)p 0.00( 0.00%)
1.0000 0.0002 0.0000 0.0000 -0.0002
0.0000 0.0000 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.0000 0.0000
18. (1.99913) CR ( 1) C 3 s(100.00%)p 0.00( 0.00%)
1.0000 0.0002 0.0000 0.0000 -0.0001
0.0000 -0.0002 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.0000 0.0000
19. (1.99918) CR ( 1) C 4 s(100.00%)p 0.00( 0.00%)
1.0000 0.0003 0.0000 0.0000 0.0002
0.0000 -0.0002 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.0000 0.0000
20. (1.99918) CR ( 1) C 5 s(100.00%)p 0.00( 0.00%)
1.0000 0.0003 0.0000 0.0000 0.0002
0.0000 0.0002 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.0000 0.0000
21. (1.99937) CR ( 1) N 12 s(100.00%)p 0.00( 0.00%)
1.0000 0.0002 0.0000 0.0000 0.0001
0.0000 0.0000 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.0000 0.0000
22. (0.00470) RY*( 1) C 1 s( 0.82%)p99.99( 93.00%)d 7.56( 6.18%)
0.0000 -0.0121 0.0895 0.0030 -0.0091
-0.5676 0.0329 0.7789 0.0000 0.0000
0.1934 0.0000 0.0000 0.1551 0.0192
23. (0.00237) RY*( 2) C 1 s( 0.07%)p99.99( 99.16%)d10.35( 0.77%)
0.0000 -0.0020 0.0203 -0.0180 -0.0387
0.8040 -0.0137 0.5861 0.0000 0.0000
0.0492 0.0000 0.0000 -0.0721 -0.0077
24. (0.00036) RY*( 3) C 1 s( 0.00%)p 1.00( 0.66%)d99.99( 99.34%)
0.0000 0.0000 0.0001 0.0000 0.0000
0.0000 0.0000 0.0000 -0.0065 0.0809
0.0000 0.8278 0.5551 0.0000 0.0000
25. (0.00023) RY*( 4) C 1 s( 86.07%)p 0.00( 0.27%)d 0.16( 13.67%)
0.0000 0.0025 0.9252 0.0688 0.0216
-0.0231 -0.0134 -0.0385 0.0000 -0.0003
-0.0445 -0.0001 0.0000 -0.3670 0.0016
26. (0.00020) RY*( 5) C 1 s( 0.00%)p 1.00( 84.50%)d 0.18( 15.50%)
0.0000 0.0000 0.0003 0.0000 0.0000
0.0000 0.0000 0.0000 -0.0025 0.9193
0.0000 0.1533 -0.3626 -0.0001 0.0000
27. (0.00007) RY*( 6) C 1 s( 12.03%)p 0.14( 1.66%)d 7.18( 86.31%)
28. (0.00004) RY*( 7) C 1 s( 36.10%)p 0.03( 1.20%)d 1.74( 62.70%)
29. (0.00000) RY*( 8) C 1 s( 0.00%)p 1.00( 14.90%)d 5.71( 85.10%)
30. (0.00001) RY*( 9) C 1 s( 9.45%)p 0.50( 4.76%)d 9.07( 85.79%)
The following information from the .log file (up to the LUMO).
Natural Bond Orbitals (Summary):
Principal Delocalizations
NBO Occupancy Energy (geminal,vicinal,remote)
====================================================================================
Molecular unit 1 (C5H6N)
1. BD ( 1) C 1 - C 2 1.98249 -0.90377 118(v),114(v),107(g),63(v),110(g),109(g),43(v),42(v),112(g)
2. BD ( 1) C 1 - C 5 1.98297 -0.92650 120(v),112(v),106(g),33(v),118(g),97(v),109(g),32(v),119(g),96(v)
3. BD ( 2) C 1 - C 5 1.61445 -0.46666 111(v),117(v),99(v),108(g),37(v)
4. BD ( 1) C 1 - H 6 1.97822 -0.71855 119(v),110(v),32(v),62(v),106(g),107(g)
5. BD ( 1) C 2 - C 3 1.98249 -0.90379 115(v),109(v),113(g),54(v),106(g),114(g),23(v),22(v),112(g)
6. BD ( 2) C 2 - C 3 1.54879 -0.44891 117(v),108(v),56(v),26(v)
7. BD ( 1) C 2 - H 7 1.98141 -0.71806 113(v),107(v),22(v),42(v),110(g),106(g)
8. BD ( 1) C 3 - C 4 1.98297 -0.92648 120(v),112(v),110(g),33(v),115(g),97(v),114(g),32(v),116(g),96(v)
9. BD ( 1) C 3 - H 8 1.97822 -0.71855 116(v),106(v),32(v),52(v),110(g),113(g)
10. BD ( 1) C 4 - H 9 1.98154 -0.75115 119(v),110(v),42(v),113(g),96(v)
11. BD ( 1) C 4 - N 12 1.98861 -1.06562 119(g),62(v),114(v),43(v),118(v),63(v),113(g),120(g)
12. BD ( 2) C 4 - N 12 1.82447 -0.56812 108(v),111(v),64(v),46(v)
13. BD ( 1) C 5 - H 11 1.98154 -0.75115 116(v),106(v),22(v),107(g),96(v)
14. BD ( 1) C 5 - N 12 1.98861 -1.06559 116(g),52(v),109(v),23(v),115(v),54(v),107(g),120(g)
15. BD ( 1) H 10 - N 12 1.98630 -0.89229 113(v),107(v),52(v),62(v)
16. CR ( 1) C 1 1.99913 -10.26482 33(v),63(v),62(v),118(v),110(v),72(v),112(v),119(v)
17. CR ( 1) C 2 1.99914 -10.27397 43(v),23(v),107(v),113(v),114(v),109(v),76(v)
18. CR ( 1) C 3 1.99913 -10.26482 33(v),54(v),52(v),115(v),106(v),80(v),112(v),116(v)
19. CR ( 1) C 4 1.99918 -10.32333 43(v),119(v),120(v),110(v),113(g),97(v),84(v),114(v)
20. CR ( 1) C 5 1.99918 -10.32332 23(v),116(v),120(v),106(v),107(g),97(v),92(v),109(v)
21. CR ( 1) N 12 1.99937 -14.46220 54(v),63(v)
22. RY*( 1) C 1 0.00470 1.06703
23. RY*( 2) C 1 0.00237 0.51525
24. RY*( 3) C 1 0.00036 1.72438
25. RY*( 4) C 1 0.00023 1.06874
26. RY*( 5) C 1 0.00020 0.58755
27. RY*( 6) C 1 0.00007 2.11820
28. RY*( 7) C 1 0.00004 2.91470
29. RY*( 8) C 1 0.00000 1.53581
30. RY*( 9) C 1 0.00001 2.30967
Borazine
Borazine is an analogue of benzene, with alternating Bs and Ns instead of Cs.
Optimization
The method was DFT/B3LYP with basis set 6-31G(d,p). This file was submitted to D-space: [15]

| X1-H length | H-X1-X2 angle | |
|---|---|---|
| X1 = B, X2 = N |  |

|
| 1.20Å | 121.4° | |
| X1 = N, X2 = B |  |

|
| 1.01Å | 118.6° |
B-N bond length is 1.43Å
The following information from the .log file shows that the parameters converged:
Item Value Threshold Converged?
Maximum Force 0.000179 0.000450 YES
RMS Force 0.000079 0.000300 YES
Maximum Displacement 0.000803 0.001800 YES
RMS Displacement 0.000300 0.001200 YES
Predicted change in Energy=-8.717126D-07
Optimization completed.
-- Stationary point found.
----------------------------
! Optimized Parameters !
! (Angstroms and Degrees) !
-------------------------- --------------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------------------
! R1 R(1,5) 1.0096 -DE/DX = 0.0 !
! R2 R(2,6) 1.0096 -DE/DX = 0.0 !
! R3 R(3,4) 1.0096 -DE/DX = 0.0 !
! R4 R(4,7) 1.4306 -DE/DX = 0.0 !
! R5 R(4,9) 1.4306 -DE/DX = 0.0 !
! R6 R(5,9) 1.4306 -DE/DX = 0.0 !
! R7 R(5,11) 1.4306 -DE/DX = 0.0 !
! R8 R(6,7) 1.4306 -DE/DX = 0.0 !
! R9 R(6,11) 1.4305 -DE/DX = 0.0 !
! R10 R(7,8) 1.1955 -DE/DX = -0.0001 !
! R11 R(9,10) 1.1955 -DE/DX = -0.0001 !
! R12 R(11,12) 1.1955 -DE/DX = -0.0001 !
! A1 A(3,4,7) 118.6037 -DE/DX = -0.0001 !
! A2 A(3,4,9) 118.5882 -DE/DX = -0.0001 !
! A3 A(7,4,9) 122.8081 -DE/DX = 0.0002 !
! A4 A(1,5,9) 118.6084 -DE/DX = -0.0001 !
! A5 A(1,5,11) 118.5849 -DE/DX = -0.0001 !
! A6 A(9,5,11) 122.8066 -DE/DX = 0.0002 !
! A7 A(2,6,7) 118.5927 -DE/DX = -0.0001 !
! A8 A(2,6,11) 118.5993 -DE/DX = -0.0001 !
! A9 A(7,6,11) 122.8081 -DE/DX = 0.0002 !
! A10 A(4,7,6) 117.1912 -DE/DX = -0.0002 !
! A11 A(4,7,8) 121.3995 -DE/DX = 0.0001 !
! A12 A(6,7,8) 121.4093 -DE/DX = 0.0001 !
! A13 A(4,9,5) 117.192 -DE/DX = -0.0002 !
! A14 A(4,9,10) 121.4067 -DE/DX = 0.0001 !
! A15 A(5,9,10) 121.4013 -DE/DX = 0.0001 !
! A16 A(5,11,6) 117.194 -DE/DX = -0.0002 !
! A17 A(5,11,12) 121.4065 -DE/DX = 0.0001 !
! A18 A(6,11,12) 121.3995 -DE/DX = 0.0001 !
! D1 D(3,4,7,6) 179.9997 -DE/DX = 0.0 !
! D2 D(3,4,7,8) -0.0012 -DE/DX = 0.0 !
! D3 D(9,4,7,6) 0.0032 -DE/DX = 0.0 !
! D4 D(9,4,7,8) -179.9977 -DE/DX = 0.0 !
! D5 D(3,4,9,5) 179.9953 -DE/DX = 0.0 !
! D6 D(3,4,9,10) -0.0066 -DE/DX = 0.0 !
! D7 D(7,4,9,5) -0.0081 -DE/DX = 0.0 !
! D8 D(7,4,9,10) 179.99 -DE/DX = 0.0 !
! D9 D(1,5,9,4) -179.9947 -DE/DX = 0.0 !
! D10 D(1,5,9,10) 0.0072 -DE/DX = 0.0 !
! D11 D(11,5,9,4) 0.0029 -DE/DX = 0.0 !
! D12 D(11,5,9,10) -179.9952 -DE/DX = 0.0 !
! D13 D(1,5,11,6) -179.9956 -DE/DX = 0.0 !
! D14 D(1,5,11,12) 0.0076 -DE/DX = 0.0 !
! D15 D(9,5,11,6) 0.0068 -DE/DX = 0.0 !
! D16 D(9,5,11,12) -179.99 -DE/DX = 0.0 !
! D17 D(2,6,7,4) 180.0001 -DE/DX = 0.0 !
! D18 D(2,6,7,8) 0.001 -DE/DX = 0.0 !
! D19 D(11,6,7,4) 0.0074 -DE/DX = 0.0 !
! D20 D(11,6,7,8) -179.9917 -DE/DX = 0.0 !
! D21 D(2,6,11,5) 179.9951 -DE/DX = 0.0 !
! D22 D(2,6,11,12) -0.0081 -DE/DX = 0.0 !
! D23 D(7,6,11,5) -0.0122 -DE/DX = 0.0 !
! D24 D(7,6,11,12) 179.9846 -DE/DX = 0.0 !
--------------------------------------------------------------------------------
GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
The data above shows that the optimized borazine molecule has B-H bond lengths of 1.20Å, H-B-N bond angles of 121.4°, N-H bond lengths of 1.01Å, H-N-B bond angles of 118.6°, and B-N bond lengths of 1.43Å. The symmetry group is C1 and has a 0.00 Debye dipole moment as it is symmetric. The job took 4 min 7.5 seconds to run. The final energy is -242.65861161 a.u.. The frequency analysis is run next to determine if the minima was found.
Frequency Analysis
The method and basis set were the same, with job = frequency. This frequency analysis was submitted to D-space: [16].

| X1-H length | H-X1-X2 angle | |
|---|---|---|
| X1 = B, X2 = N |  |

|
| 1.20Å | 121.4° | |
| X1 = N, X2 = B |  |

|
| 1.01Å | 118.6° |
B-N bond length is 1.43Å.
The frequency calculation was successful, and the following data shows that all the parameters converged:
Item Value Threshold Converged? Maximum Force 0.000475 0.000450 YES RMS Force 0.000176 0.000300 YES Maximum Displacement 0.000857 0.001800 YES RMS Displacement 0.000413 0.001200 YES Predicted change in Energy=-9.713623D-07 GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
Low frequencies --- -6.0015 -0.0006 0.0006 0.0006 8.5529 12.9759 Low frequencies --- 288.7601 290.6694 404.2361
Vibrational analysis
| Mode | Frequency (cm-1) | Intensity | Mode | Frequency (cm-1) | Intensity | Mode | Frequency (cm-1) | Intensity |
|---|---|---|---|---|---|---|---|---|
| 1 | 289 | 0 | 11 | 929 | 0 | 21 | 1400 | 10 |
| 2 | 291 | 0 | 12 | 938 | 235 | 22 | 1400 | 10 |
| 3 | 404 | 24 | 13 | 945 | 0 | 23 | 1492 | 495 |
| 4 | 525 | 1 | 14 | 945 | 0 | 24 | 1492 | 495 |
| 5 | 525 | 1 | 15 | 945 | 0 | 25 | 2637 | 284 |
| 6 | 708 | 0 | 16 | 1053 | 0 | 26 | 2637 | 284 |
| 7 | 710 | 0 | 17 | 1081 | 0 | 27 | 2647 | 0 |
| 8 | 732 | 61 | 18 | 1081 | 0 | 28 | 3643 | 0 |
| 9 | 864 | 0 | 19 | 1246 | 0 | 29 | 3645 | 40 |
| 10 | 929 | 0 | 20 | 1314 | 0 | 30 | 3645 | 40 |
This gives the following IR spectrum:

MO analysis
The same method and basis set were used, job = energy. The link to D-space can be found here: [17]
There is degeneracy again in this molecule. The energy of the HOMO is -0.27599 a.u. and the energy of the LUMO is 0.02422 a.u..
Charge distribution
An NBO analysis was run on the .log file of the optimized borazine molecule. The color range was from -1.250 to +1.250.


The relative charges were 0.432 for the H attached to N, -0.077 for the H attached to B, 0.747 for B, and -1.103 for N.
The following part of the .log file shows the natural charge information:
Summary of Natural Population Analysis:
Natural Population
Natural -----------------------------------------------
Atom No Charge Core Valence Rydberg Total
-----------------------------------------------------------------------
H 1 0.43212 0.00000 0.56560 0.00228 0.56788
H 2 0.43213 0.00000 0.56559 0.00228 0.56787
H 3 0.43213 0.00000 0.56559 0.00228 0.56787
N 4 -1.10251 1.99943 6.09830 0.00478 8.10251
N 5 -1.10250 1.99943 6.09829 0.00478 8.10250
N 6 -1.10251 1.99943 6.09830 0.00478 8.10251
B 7 0.74698 1.99917 2.23862 0.01522 4.25302
H 8 -0.07657 0.00000 1.07588 0.00069 1.07657
B 9 0.74697 1.99917 2.23863 0.01522 4.25303
H 10 -0.07657 0.00000 1.07588 0.00069 1.07657
B 11 0.74691 1.99917 2.23870 0.01523 4.25309
H 12 -0.07657 0.00000 1.07588 0.00069 1.07657
=======================================================================
* Total * 0.00000 11.99579 29.93527 0.06894 42.00000
The following information from the .log file (up to the LUMO) gives precise details on electron density.
(Occupancy) Bond orbital/ Coefficients/ Hybrids
---------------------------------------------------------------------------------
1. (1.98494) BD ( 1) H 1 - N 5
( 28.07%) 0.5298* H 1 s( 99.91%)p 0.00( 0.09%)
0.9996 -0.0010 -0.0145 -0.0257 0.0000
( 71.93%) 0.8481* N 5 s( 22.85%)p 3.37( 77.12%)d 0.00( 0.03%)
-0.0002 0.4779 -0.0114 0.0006 0.4307
0.0064 0.7652 0.0114 0.0000 0.0000
0.0104 0.0000 0.0000 -0.0063 -0.0119
2. (1.98494) BD ( 1) H 2 - N 6
( 28.07%) 0.5298* H 2 s( 99.91%)p 0.00( 0.09%)
-0.9996 0.0010 0.0150 -0.0254 0.0000
( 71.93%) 0.8481* N 6 s( 22.85%)p 3.37( 77.12%)d 0.00( 0.03%)
0.0002 -0.4779 0.0114 -0.0006 -0.4474
-0.0066 0.7556 0.0112 0.0000 0.0000
0.0107 0.0000 0.0000 0.0058 0.0119
3. (1.98494) BD ( 1) H 3 - N 4
( 28.07%) 0.5298* H 3 s( 99.91%)p 0.00( 0.09%)
-0.9996 0.0010 -0.0295 -0.0003 0.0000
( 71.93%) 0.8481* N 4 s( 22.85%)p 3.37( 77.12%)d 0.00( 0.03%)
0.0002 -0.4779 0.0114 -0.0006 0.8780
0.0130 0.0096 0.0001 0.0000 0.0000
-0.0003 0.0000 0.0000 -0.0122 0.0119
4. (1.98437) BD ( 1) N 4 - B 7
( 76.47%) 0.8745* N 4 s( 38.54%)p 1.59( 61.45%)d 0.00( 0.01%)
0.0000 -0.6208 -0.0043 0.0001 -0.3458
0.0160 0.7033 0.0001 0.0000 0.0000
0.0058 0.0000 0.0000 0.0042 0.0085
( 23.53%) 0.4851* B 7 s( 31.27%)p 2.19( 68.49%)d 0.01( 0.25%)
0.0003 -0.5589 0.0174 -0.0032 0.4013
0.0541 -0.7214 -0.0207 -0.0001 0.0000
0.0366 0.0000 0.0000 0.0265 0.0206
5. (1.82091) BD ( 2) N 4 - B 7
( 88.21%) 0.9392* N 4 s( 0.00%)p 1.00(100.00%)d 0.00( 0.00%)
0.0000 0.0000 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 1.0000 -0.0003
0.0000 0.0047 -0.0004 0.0000 0.0000
( 11.79%) 0.3433* B 7 s( 0.00%)p 1.00( 99.62%)d 0.00( 0.38%)
0.0000 0.0000 0.0000 0.0000 0.0001
0.0000 0.0000 0.0000 0.9976 -0.0314
0.0000 -0.0227 0.0571 0.0000 0.0000
6. (1.98436) BD ( 1) N 4 - B 9
( 76.47%) 0.8745* N 4 s( 38.53%)p 1.59( 61.46%)d 0.00( 0.01%)
0.0000 0.6207 0.0043 -0.0001 0.3303
-0.0159 0.7108 -0.0003 0.0000 0.0000
0.0056 0.0000 0.0000 -0.0045 -0.0085
( 23.53%) 0.4851* B 9 s( 31.27%)p 2.19( 68.48%)d 0.01( 0.25%)
-0.0003 0.5589 -0.0174 0.0032 -0.3853
-0.0536 -0.7301 -0.0218 0.0000 0.0000
0.0354 0.0000 0.0000 -0.0281 -0.0206
7. (1.98437) BD ( 1) N 5 - B 9
( 76.47%) 0.8745* N 5 s( 38.54%)p 1.59( 61.45%)d 0.00( 0.01%)
0.0000 -0.6208 -0.0043 0.0001 0.7820
-0.0079 -0.0522 -0.0139 0.0001 0.0000
0.0007 0.0000 0.0000 -0.0072 0.0085
( 23.53%) 0.4851* B 9 s( 31.27%)p 2.19( 68.49%)d 0.01( 0.25%)
0.0003 -0.5589 0.0174 -0.0032 -0.8254
-0.0449 0.0131 -0.0365 0.0000 0.0000
0.0046 0.0000 0.0000 -0.0450 0.0206
8. (1.82090) BD ( 2) N 5 - B 9
( 88.21%) 0.9392* N 5 s( 0.00%)p 1.00(100.00%)d 0.00( 0.00%)
0.0000 0.0000 0.0000 0.0000 -0.0001
0.0000 0.0000 0.0000 1.0000 -0.0003
0.0000 -0.0027 -0.0038 0.0000 0.0000
( 11.79%) 0.3433* B 9 s( 0.00%)p 1.00( 99.62%)d 0.00( 0.38%)
0.0000 0.0000 0.0000 0.0000 0.0000
0.0000 0.0001 0.0000 0.9976 -0.0314
0.0000 0.0608 -0.0089 0.0000 0.0000
9. (1.98436) BD ( 1) N 5 - B 11
( 76.47%) 0.8745* N 5 s( 38.53%)p 1.59( 61.45%)d 0.00( 0.01%)
0.0000 0.6207 0.0043 -0.0001 0.4504
0.0078 -0.6414 0.0139 0.0000 0.0000
-0.0067 0.0000 0.0000 -0.0026 -0.0085
( 23.53%) 0.4851* B 11 s( 31.27%)p 2.19( 68.48%)d 0.01( 0.25%)
-0.0003 0.5589 -0.0174 0.0032 -0.4396
0.0079 0.6987 0.0574 0.0000 0.0000
-0.0420 0.0000 0.0000 -0.0166 -0.0206
10. (1.98436) BD ( 1) N 6 - B 7
( 76.47%) 0.8745* N 6 s( 38.53%)p 1.60( 61.46%)d 0.00( 0.01%)
0.0000 -0.6207 -0.0044 0.0001 0.7807
-0.0082 0.0694 0.0137 0.0000 0.0000
-0.0011 0.0000 0.0000 -0.0071 0.0085
( 23.53%) 0.4851* B 7 s( 31.27%)p 2.19( 68.48%)d 0.01( 0.25%)
0.0003 -0.5589 0.0174 -0.0032 -0.8249
-0.0457 -0.0313 0.0355 0.0001 0.0000
-0.0066 0.0000 0.0000 -0.0447 0.0206
11. (1.98436) BD ( 1) N 6 - B 11
( 76.47%) 0.8745* N 6 s( 38.54%)p 1.59( 61.45%)d 0.00( 0.01%)
0.0000 0.6208 0.0043 -0.0001 0.4361
0.0081 0.6512 -0.0138 0.0000 0.0000
0.0066 0.0000 0.0000 -0.0029 -0.0085
( 23.53%) 0.4851* B 11 s( 31.27%)p 2.19( 68.49%)d 0.01( 0.25%)
-0.0003 0.5589 -0.0174 0.0032 -0.4241
0.0092 -0.7083 -0.0572 -0.0001 0.0000
0.0413 0.0000 0.0000 -0.0185 -0.0206
12. (1.82094) BD ( 2) N 6 - B 11
( 88.21%) 0.9392* N 6 s( 0.00%)p 1.00(100.00%)d 0.00( 0.00%)
0.0000 0.0000 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 1.0000 -0.0003
0.0000 -0.0019 0.0043 0.0000 0.0000
( 11.79%) 0.3433* B 11 s( 0.00%)p 1.00( 99.62%)d 0.00( 0.38%)
0.0000 0.0000 0.0000 0.0000 -0.0001
0.0000 -0.0001 0.0000 0.9976 -0.0314
0.0000 -0.0381 -0.0482 0.0000 0.0000
13. (1.98669) BD ( 1) B 7 - H 8
( 45.97%) 0.6780* B 7 s( 37.44%)p 1.67( 62.50%)d 0.00( 0.07%)
-0.0006 0.6117 0.0129 -0.0016 -0.3875
0.0132 -0.6886 0.0235 0.0000 0.0000
0.0202 0.0000 0.0000 -0.0122 -0.0098
( 54.03%) 0.7351* H 8 s( 99.96%)p 0.00( 0.04%)
0.9998 0.0002 0.0094 0.0167 0.0000
14. (1.98669) BD ( 1) B 9 - H 10
( 45.97%) 0.6780* B 9 s( 37.44%)p 1.67( 62.50%)d 0.00( 0.07%)
-0.0006 0.6117 0.0129 -0.0016 -0.4026
0.0137 0.6798 -0.0232 -0.0001 0.0000
-0.0207 0.0000 0.0000 -0.0114 -0.0098
( 54.03%) 0.7351* H 10 s( 99.96%)p 0.00( 0.04%)
0.9998 0.0002 0.0098 -0.0165 0.0000
15. (1.98668) BD ( 1) B 11 - H 12
( 45.97%) 0.6780* B 11 s( 37.44%)p 1.67( 62.50%)d 0.00( 0.07%)
-0.0006 0.6117 0.0129 -0.0016 0.7901
-0.0270 0.0087 -0.0003 0.0001 0.0000
0.0005 0.0000 0.0000 0.0236 -0.0098
( 54.03%) 0.7351* H 12 s( 99.96%)p 0.00( 0.04%)
0.9998 0.0002 -0.0192 -0.0002 0.0000
16. (1.99943) CR ( 1) N 4 s(100.00%)p 0.00( 0.00%)
1.0000 0.0001 0.0000 0.0000 -0.0002
0.0000 0.0000 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.0000 0.0000
17. (1.99943) CR ( 1) N 5 s(100.00%)p 0.00( 0.00%)
1.0000 0.0001 0.0000 0.0000 0.0001
0.0000 0.0001 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.0000 0.0000
18. (1.99943) CR ( 1) N 6 s(100.00%)p 0.00( 0.00%)
1.0000 0.0001 0.0000 0.0000 0.0001
0.0000 -0.0001 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.0000 0.0000
19. (1.99917) CR ( 1) B 7 s(100.00%)p 0.00( 0.00%)
1.0000 0.0008 0.0000 0.0000 -0.0001
0.0000 -0.0002 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.0000 0.0000
20. (1.99917) CR ( 1) B 9 s(100.00%)p 0.00( 0.00%)
1.0000 0.0008 0.0000 0.0000 -0.0001
0.0000 0.0002 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.0000 0.0000
21. (1.99917) CR ( 1) B 11 s(100.00%)p 0.00( 0.00%)
1.0000 0.0008 0.0000 0.0000 0.0002
0.0000 0.0000 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.0000 0.0000
22. (0.00102) RY*( 1) H 1 s( 98.57%)p 0.01( 1.43%)
0.0046 0.9928 0.0583 0.1044 0.0000
The following information from the .log file (up to the LUMO).
Natural Bond Orbitals (Summary):
Principal Delocalizations
NBO Occupancy Energy (geminal,vicinal,remote)
====================================================================================
Molecular unit 1 (H6B3N3)
1. BD ( 1) H 1 - N 5 1.98494 -0.61506 116(v),111(v),114(g),112(g),92(v),78(v)
2. BD ( 1) H 2 - N 6 1.98494 -0.61507 114(v),109(v),116(g),115(g),64(v),92(v)
3. BD ( 1) H 3 - N 4 1.98494 -0.61508 112(v),115(v),109(g),111(g),78(v),64(v)
4. BD ( 1) N 4 - B 7 1.98437 -0.68880 111(g),107(v),108(g),119(v),79(v),112(v)
5. BD ( 2) N 4 - B 7 1.82091 -0.27149 113(v),84(v),80(v),31(v),110(g)
6. BD ( 1) N 4 - B 9 1.98436 -0.68877 109(g),106(v),108(g),118(v),65(v),115(v)
7. BD ( 1) N 5 - B 9 1.98437 -0.68876 114(g),108(v),106(g),120(v),93(v),116(v)
8. BD ( 2) N 5 - B 9 1.82090 -0.27146 117(v),98(v),94(v),23(v),113(g)
9. BD ( 1) N 5 - B 11 1.98436 -0.68876 112(g),107(v),106(g),119(v),79(v),111(v)
10. BD ( 1) N 6 - B 7 1.98436 -0.68872 116(g),108(v),107(g),120(v),93(v),114(v)
11. BD ( 1) N 6 - B 11 1.98436 -0.68882 115(g),106(v),107(g),118(v),65(v),109(v)
12. BD ( 2) N 6 - B 11 1.82094 -0.27148 110(v),70(v),66(v),27(v),117(g)
13. BD ( 1) B 7 - H 8 1.98669 -0.40371 111(v),116(v),34(v),54(v)
14. BD ( 1) B 9 - H 10 1.98669 -0.40370 109(v),114(v),44(v),34(v)
15. BD ( 1) B 11 - H 12 1.98668 -0.40369 115(v),112(v),54(v),44(v)
16. CR ( 1) N 4 1.99943 -14.13100 65(v),79(v),109(g),111(g)
17. CR ( 1) N 5 1.99943 -14.13099 79(v),93(v),114(g),112(g)
18. CR ( 1) N 6 1.99943 -14.13099 93(v),65(v),116(g),115(g)
19. CR ( 1) B 7 1.99917 -6.65255 111(v),116(v),108(v),107(v)
20. CR ( 1) B 9 1.99917 -6.65254 109(v),114(v),106(v),108(v)
21. CR ( 1) B 11 1.99917 -6.65251 115(v),112(v),107(v),106(v)
22. RY*( 1) H 1 0.00102 0.69891
Comparison of charge distribution
| Benzene | Boratabenzene | Pyridinium | Borazine |
|---|---|---|---|
|
|
|
|
|
|
|
|
The N in borazine, which is bonded to two Bs, is more negatively charged (-1.103) than the N in pyridiunium, which is bonded to two Cs (-0.476). This is because B is more electropositive than C, thus the electron density is pushed towards the N for borazine.
The ortho-C atoms in pyridinium are more positively charged (0.071) than the meta-C atoms (-0.241), and the para-C (-0.122). The para-C is more positively charged than the meta-Cs, which could indicate that the N is withdrawing electron density across the aromatic ring. This is also true for boratabenzene, where the all the Cs except for para-C are less negatively charged (-0.108) than para-C (-0.113).
For the H attached to B in boratabenzene, it has a higher negative charge (-0.224) than all the other Hs attached to C. For the H attached to N in pyridinium, it has a higher positive charge (0.483) than all the other Hs attached to C. For borazine, the H attached to N is highly positive (0.432) in comparison to the H attached to B (-0.077). In these cases, N is withdrawing electron density and thus gives the H a more positive value, whereas B is donating electron density.
Benzene has the lowest range due to it not having any heteroatoms in the ring, and having no overall dipole moment due to its symmetry. Borazine has the highest range due to the fact that B and N have the highest electronegativity difference.
Comparison of MOs
| MO | Benzene | Boratabenzene | Pyridinium | Borazine |
|---|---|---|---|---|
| pi totally bonding |  |
 |
 |

|
| LCAO |  |
 |
 |

|
| Energy | -0.35998 | -0.13207 | -0.64074 | -0.36138 |
All four of these MOs have a delocalized electron ring above and below the plane of the molecule. Each shows aromaticity: benzene has 6 pi electrons, boratabenzene has a -1 charge that contributes to the ring, pyridinium has a N lone pair that is not part of the aromatic ring, and borazine has the correct number of electrons to be considered aromatic. All four MOs have one nodal plane that is in the plane of the ring. Looking at the fact that it is totally bonding, this is to be excpected.
Pyridinium has the lowest energy due to the N lowering the energy of most of the MOs as it is more electronegative than C. Boratabenzene has the highest energy due to the B which is highly electropositive, thus raising the energies of the MOs. Borazine has both N and B atoms thus they pretty much level out and the energy is similar to that of benzene. For all of the MOs, pyridinium has the lowest energy, followed by benzene or borazine, and then followed by boratabenzene which has the highest energy.
For the pyridinium MO, it can be see that there is a higher charge density towards the N, which occurs as it withdraws the electron density from the other atoms. The opposite is true for boratabenzne, which has a smaller electron density near the B. Both benzene and borazine are symmetric and thus have symmetrical MOs.
The LCAOs were drawn showing the electronegativity difference between C, B, and N.
| MO | Benzene | Boratabenzene | Pyridinium | Borazine |
|---|---|---|---|---|
| HOMO |  |
 |
 |

|
| LCAO |  |
 |
 |

|
| Energy | -0.24691 | 0.01095 | -0.47885 | -0.27599 |
| MO | Benzene | Boratabenzene | Pyridinium | Borazine |
|---|---|---|---|---|
| HOMO-1 |  |
 |
 |

|
| LCAO |  |
 |
 |

|
| Energy | -0.24692 | -0.03492 | -0.50847 | -0.27601 |
For both the HOMO and HOMO-1, there are two nodal planes: one in the plane of the ring, and one perpendicular to it in the middle. Since benzene and borazine are both symmetrical molecules, the HOMO and HOMO-1 MOs are doubly degenerate. This is not the case for boratazene or pyridinium, neither of which are symmetrical.
Pyridinium's HOMO has a node at the N, whereas its HOMO-1 MO has electron density on the N. This is results in the HOMO-1 being lower in energy in comparison to the HOMO. The opposite is true for boratabenzene which has a node at the B in its HOMO-1, and electron density on its B for its HOMO MO. Since B is electropositive it is destabilized with increasing electron density. Thus the HOMO has the higher energy due to the fact that there is a -1 charge on the B.
The effect of these substitutions of benzene to create these analogues affects the relative energies of the AOs and thus the MO diagram. N has a higher electronegativity than C, and thus lowers all the levels of the MOs. B has a lower electronegativity and thus raises the energies of all the levels.
References
- ↑ 1.0 1.1 M. Schuurman, et. al., J. Comp . Chem., 26, 2005, pp.1106-1112
- ↑ 2.0 2.1 P. Hunt, MO tutorial year 2 [1]
- ↑ J. Glaser and G. Johansson, Acta Chemica Scandinavica 1982, 36a, 125-135
- ↑ J. E. D. Davies et al., J. Chem. Soc., 1968, 2050
- ↑ Z. Anorg. Allg. Chem. 2008, 704-707
- ↑ S. Xie, Y. Song, Z. Liu, Can. J. Chem., 87, 2009, 1235-1247
- ↑ Yu. Kh. Shaulov; G. O. Shmyreva; V. S. Tubyanskaya, Zhurnal Fizicheskoi Khimii, 1966, 40, 122-124.