
Rep:Mod:physMALIX9093
The Cope Rearrangement
The first part of this project involved studying the chemical reactivity of the Cope rearrangement of 1,5-hexadiene. More specifically the low-energy minima and 'chair' and 'boat' transition structures were located on the C6H10 potential energy surface, using GaussView 5.0, to determine the preferred reaction mechanism.
Optimisation the Reactants and Products
Optimisation of 1,5-hexadiene [Anti1]
| Molecule | 
|
| Links to the output files | log/ chk |
| Calculation Type | FOPT |
| Calculation Method | RHF |
| Basis set | 3-21G |
| Final Energy (a.u.) | -231.69260220 |
| RMS Gradient Norm (a.u.) | 0.00004706 |
| Dipole Moment (Debye) | 0.2027 |
| Point Group | C2 |
| Time of calculation (s) | 23 |
A molecule of 1,5-hexadiene with an 'anti' linkage for the four central carbons atoms was created using GaussView 5.0. The molecule was cleaned before it was optimised using the Hartree Fock method and 3-21G basis set. 3-21G basis set is a low level basis set, with very low level of accuracy, but has the advantage of leading to fast calculation times[1]. The molecule was symmetrised; this gave the C2 point group. A summary table containing all the information regarding the calculation, including links to the output files, can be found here on the left.
The RMS gradient was found to be close to zero (in fact less than 0.001), 0.00004706 a.u., which means that the molecule was successfully optimised[1]This was further confirmed by looking at the convergence of the forces and displacements in the output files, as shown here below.
Item Value Threshold Converged? Maximum Force 0.000120 0.000450 YES RMS Force 0.000024 0.000300 YES Maximum Displacement 0.001357 0.001800 YES RMS Displacement 0.000425 0.001200 YES Predicted change in Energy=-1.688879D-07 Optimization completed. -- Stationary point found.
Optimisation of 1,5-hexadiene [Gauche 5]
| Molecule | 
|
| Link to output files | log/ chk |
| Calculation Type | FOPT |
| Calculation Method | RHF |
| Basis set | 3-21G |
| Final Energy (a.u.) | -231.68961573 |
| RMS Gradient Norm(a.u.) | 0.00000942 |
| Dipole Moment(Debye) | 0.4433 |
| Point Group (after symmetrisation) | C1 |
| Time of calculation | 10 min 19 s |
Another molecule of 1,5-hexadiene was created but this time, with a 'gauche' linkage for the four central carbon atoms.
Would you expect this structure to have a lower or a higher energy than the anti structure you have just optimised?
Gauche and antiperiplanar conformations correspond to staggered conformations and hence energy minima, while eclipsed conformations correspond to energy maxima[2]. To compare the relative stabilities of gauche and antiperiplanar conformations, two major effects need to be considered: the effect of stabilising σ C-H / σ* C-H and σ C-C / σ* C-C orbital interactions and the Pauli repulsion energy [2] which both favour the antiperiplanar conformation in the case of n-butane, for example (as a result n-butane shows a difference of energy of -0.8 kcal/mol between the anti and gauche forms[3]). In the gauche form, proximal hydrogens are seperated by 2.35 A [3], in the case of n-butane, while vinyl hydrogens are seperated by 2.5 A[3], in the case of 1,5-hexadiene. Therefore, in the case 1,5-hexadiene, hydrogen atoms are not so close to each other, so repulsive steric interactions should be reduced and hence the gauche and anti conformations should be comparable in terms of stability[3].
As previously, the molecule was optimised using the Hartree Fock method and 3-21G basis set. It was then was symmetrised; this gave the C1 point group.
The RMS gradient was found to be close to zero (0.00000942 a.u.), meaning that the molecule had been successfully optimised. This was further confirmed by looking at the convergence of the forces and displacements in the output files, as shown here below.
Item Value Threshold Converged? Maximum Force 0.000020 0.000450 YES RMS Force 0.000006 0.000300 YES Maximum Displacement 0.001602 0.001800 YES RMS Displacement 0.000612 0.001200 YES Predicted change in Energy=-3.729830D-08 Optimization completed. -- Stationary point found.
The difference in energy between the anti and the gauche conformers of 1,5-hexadiene previously optmised is of 2.9865 x 10-3 a.u. i.e. 1.87 kcal/mol, which is larger than expected. This could explained by the difference in basis set chosen to run the calculations [HF/3-21G versus MP2/6-31G*[3]] and/or by the close proximity of the vinyl protons on C2 and C5. In fact, Gauche 5 is unlikely to be the lowest energy gauche conformer.
Optimisation of 1,5-hexadiene [Gauche 3]
Normally, calculated activation energies and enthalpies use the lowest energy conformation of a reactant molecule as a reference. Based on your results from above, try to predict what the lowest energy conformation of 1,5-hexadiene might be. Test out your hypothesis by drawing the structure and optimising it.
| Molecule | 
|
| Link to output files | log/ chk |
| Calculation Type | FOPT |
| Calculation Method | RHF |
| Basis set | 3-21G |
| Final Energy (a.u.) | -231.69266120 |
| RMS Gradient Norm (a.u.) | 0.00001556 |
| Dipole Moment (Debye) | 0.3406 |
| Point Group | C1 |
| Time of calculation (s) | 41 |
The hypothetic lowest energy conformer (gauche 3) was optimised using the Hartree Fock method and 3-21G basis set. It was then was symmetrised; this gave the C1 point group.
The RMS gradient was found to be close to zero (0.00001556 a.u.), meaning that the molecule had been successfully optimised. This was further confirmed by looking at the convergence of the forces and displacements in the output files, as shown here below.
Item Value Threshold Converged? Maximum Force 0.000044 0.000450 YES RMS Force 0.000009 0.000300 YES Maximum Displacement 0.001423 0.001800 YES RMS Displacement 0.000493 0.001200 YES Predicted change in Energy=-3.610688D-08 Optimization completed. -- Stationary point found.
Optimisation of 1,5-hexadiene [Anti 2]
First Optimisation
| Molecule | 
|
| Link to output files | log/ chk |
| Calculation Type | FOPT |
| Calculation Method | RHF |
| Basis set | 3-21G |
| Final Energy (a.u.) | -231.69253528 |
| RMS Gradient Norm (a.u.) | 0.00001891 |
| Dipole Moment (Debye) | 0.0000 |
| Point Group | Ci |
| Time of calculation (s) | 34.0 |
The Ci anti 2 conformer of 1,5-hexadiene was optimised using the Hartree Fock method and 3-21G basis set. The obtained molecule was then was symmetrised, which gave the Ci point group.
The RMS gradient was found to be close to zero (0.00001556 a.u.), meaning that the molecule had been successfully optimised. This was further confirmed by looking at the convergence of the forces and displacements in the output files, as shown here below.
Item Value Threshold Converged? Maximum Force 0.000060 0.000450 YES RMS Force 0.000010 0.000300 YES Maximum Displacement 0.000516 0.001800 YES RMS Displacement 0.000171 0.001200 YES Predicted change in Energy=-2.036922D-08 Optimization completed. -- Stationary point found.
Second Optimisation: Using a Higher Basis Set
| Molecule | 
|
| Link to output files | log |
| Calculation Type | FOPT |
| Calculation Method | RB3LYP |
| Basis set | 6-31G(d) |
| Final Energy (a.u.) | -234.61172154 |
| RMS Gradient Norm (a.u.) | 0.00001138 |
| Dipole Moment (Debye) | 0.0000 |
| Point Group | Ci |
| Time of calculation | 1 min 13.0 s |
The Ci anti 2 conformer of 1,5-hexadiene was then optimised using RB3LYP method and higher level 6-31G* (or 6-31G(d)) basis set, as the first calculations were run using a low level basis set (3-21G) which only provides a rough approximation of the structure.
The RMS gradient was found to be close to zero (0.00001138 a.u.), meaning that the molecule had been successfully optimised. This was further confirmed by looking at the convergence of the forces and displacements in the output files, as shown here below.
Item Value Threshold Converged? Maximum Force 0.000014 0.000450 YES RMS Force 0.000006 0.000300 YES Maximum Displacement 0.000254 0.001800 YES RMS Displacement 0.000083 0.001200 YES Predicted change in Energy=-1.337526D-08 Optimization completed. -- Stationary point found.
Now compare the final structures from the HF/3-21G calculation with that at the higher level of theory. How much does the overall geometry change?
The structure doesn't seem to have changed that much, however the energy has dropped down significantly.
Frequency Analysis of 1,5-hexadiene [Anti 2]
A frequency analysis was carried out to ensure that the optimised geometry was in its most stable ground state, rather than in the transition state[1].
After calculating the second derivative of the potential energy surface with respect to distance, the obtained frequencies are examined; if all the frequencies are positive then a minima has been found, corresponding to the ground state, but if one of the frequencies is negative, a maximum has been found, corresponding to the transition state[1].
It is essential to run the optimisation and frequency analysis using the same basis set and method otherwise some discrepancies in the energy values may arise. The optimisation of the geometry corresponds to the the first step for any quantum chemical calculation as we are determining the optimium position of the nuclei for a given electronic configuration[1]. The method used determines the type of approximations that are made in solving the Schrodinger equation, while the the basis set determines the accuracy, and the frequency analyses involves calculating the second derivative of the potential energy surface[1].
| Link to output files | log |
| Calculation Type | FREQ |
| Calculation Method | RB3LYP |
| Basis set | 6-31G(d) |
| Final Energy (a.u.) | -234.61172154 |
| RMS Gradient Norm (a.u.) | 0.00001136 |
| Dipole Moment (Debye) | 0.0000 |
| Point Group | Ci |
| Time of calculation | 1 min 23.0 s |
A frequency calculation was run on the previously optimised B3LYP/6-31G* structure.
The output file was checked in order to ensure that the forces and displacements had converged and that low frequencies were within in a small range, as shown here below.
Non-linear molecules possess 3N-6 vibrational frequencies. The 'low frequencies' represent the "-6" of the equation and represent the motions of the center of mass of the molecule[2]. These should small - in fact, the closer they are to zero, the better the optimisation - and within a range of +/- 15cm-1.[2]
Item Value Threshold Converged? Maximum Force 0.000030 0.000450 YES RMS Force 0.000011 0.000300 YES Maximum Displacement 0.000296 0.001800 YES RMS Displacement 0.000117 0.001200 YES Predicted change in Energy=-1.214787D-08 Optimization completed. -- Stationary point found.
Low frequencies --- -6.1181 0.0010 0.0014 0.0015 6.9408 22.0557 Low frequencies --- 76.6392 83.5568 122.5615
All the vibrations were inspected (they can found following this link), making sure that none of them were imaginary and the IR spectrum was simulated, as shown here below.
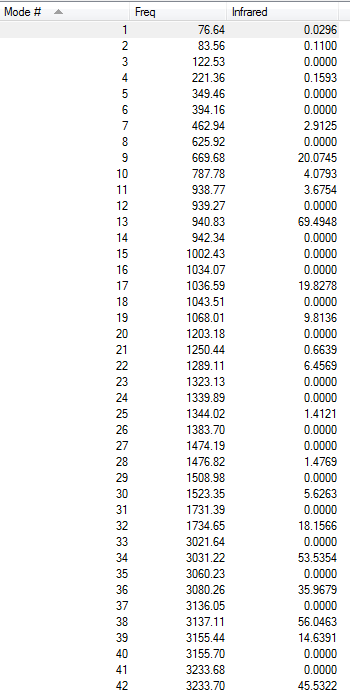{kind=link}

Energies under vibrational temperatures were recorded, as shown here below. The first value corresponds to the potential energy, at 0 K, taking into account the zero-point vibrational energy (E = Eelec + ZPE), the second one corresponds to the energy, which includes the relative contributions of the translational, rotational, and vibrational modes, under standard conditions(298.15 K and 1 atm)(E = E + Evib + Erot + Etrans), the third one contains an additional correction, which accounts for thermal energy (H = E + RT)(and is particularly important when looking at dissociation reactions), and the fourth one includes the entropic contribution to the free energy (G = H - TS)[4].
Sum of electronic and zero-point Energies= -234.469182 Sum of electronic and thermal Energies= -234.461850 Sum of electronic and thermal Enthalpies= -234.460906 Sum of electronic and thermal Free Energies= -234.500698
Note that these corrections can also be calculated at other temperatures using the Temperature option in Gaussian. If you have time, try to re-calculate these quantities at 0 K as shown in the Advanced GaussView Tutorial.
Conformations of 1,5-hexadiene
| Conformer | Structure | Links to Output Files | Point Group | Energy (a.u.) | Relative Energy (kcal/mol) |
|---|---|---|---|---|---|
| Gauche 1 |  |
log/ chk | C2 | -231.68771615 | |
| Gauche 2 |  |
log/ chk | C2 | -231.69166689 | 0.623994 |
| Gauche 3 |  |
log/ chk | C1 | -231.69266120 | 0.000000 |
| Gauche 4 |  |
log /chk | C2 | -231.69153032 | 0.709650 |
| Gauche 5 |  |
log/ chk | C1 | -231.68961573 | 1.91108 |
| Gauche 6 |  |
log/ chk | C1 | -231.68916017 | 2.19697 |
| Anti 1 |  |
log/ chk | C2 | -231.69260220 | 0.037023 |
| Anti 2 |  |
log/ chk | Ci | -231.69253528 | 0.079066 |
| Anti 3 |  |
log/ chk | C2h | -231.68907065 | |
| Anti 4 |  |
log/ chk | C1 | -231.69097054 | 1.06093 |
Optimisation of the chair and boat transition states
This section involved optimising the 'chair' and 'boat' transition state structures, in order to visualise the reaction coordinate, run the IRC (Intrinisic Reaction Coordinate) and calculate the activation energies for the Cope rearrangement.
Optimisation of an Allyl Fragment
| Molecule | 
|
| Links to output files | log/ chk |
| Calculation Type | FOPT |
| Calculation Method | UHF |
| Basis set | 3-21G |
| Final Energy (a.u.) | -115.82303994 |
| RMS Gradient Norm (a.u.) | 0.00008791 |
| Dipole Moment (Debye) | 0.0293 |
| Point Group | C1 |
| Point Group (after symmetrisation) | C2v |
| Time of calculation (s) | 24.0 |
An allyl fragment was created and then optimised using the Hartree Fock method and 3-21G basis set.
The RMS gradient was found to be close to zero (0.00008791 a.u.), meaning that the molecule had been successfully optimised. This was further confirmed by looking at the convergence of the forces and displacements in the output files, as shown here below.
Item Value Threshold Converged? Maximum Force 0.000102 0.000450 YES RMS Force 0.000049 0.000300 YES Maximum Displacement 0.000791 0.001800 YES RMS Displacement 0.000306 0.001200 YES Predicted change in Energy=-1.644639D-07 Optimization completed. -- Stationary point found.
Optimisation of the Chair Transition State
Optimisation by Computing the Hessian
The 'chair' transition state was first optimised using a method known as the Hessian. This is the preferred method as it is the easiest, but it requires to guess the transition state's geometry reasonably well. Indeed, it computs the force constant matrix in the first step of the optimisation, and the guessed structure is then revised as the optimisation proceeds[4].
| Molecule | 
|
| Links to output files | log/ chk |
| Calculation Type | FOPT |
| Calculation Method | RHF |
| Basis set | 3-21G |
| Final Energy (a.u.) | -231.61932239 |
| RMS Gradient Norm (a.u.) | 0.00003300 |
| Dipole Moment (Debye) | 0.0005 |
| Point Group | C1 |
| Point Group (after symmetrisation) | C2h |
| Time of calculation (s) | 10.0 |
The previously optimised allyl fragment was duplicated and both fragments were oriented such that they would like the 'chair' transition state, making sure that the terminal ends of the allyl fragments were seperated by approximately 2.2 Å. The transition state was then optimised using the Hartree Fock method and 3-21G basis set. To do so, the 'Optimisation' command was set to a 'TS (Berny)', force constants were only calculated once, and the additional keywords Opt=NoEigen were included, to prevent the calculation from stopping if more than one imaginary frequency is detected.
The RMS gradient was found to be close to zero (0.00008791 a.u.), meaning that the molecule had been successfully optimised. This was further confirmed by looking at the convergence of the forces and displacements in the output files, as shown here below.
Item Value Threshold Converged? Maximum Force 0.000065 0.000450 YES RMS Force 0.000015 0.000300 YES Maximum Displacement 0.001493 0.001800 YES RMS Displacement 0.000342 0.001200 YES Predicted change in Energy=-8.532989D-08 Optimization completed. -- Stationary point found.
As shown in the summary table, the obtained structure looked like a 'chair' transition state, and symmetrisation of the structure gave the C2h point group, as expected.
All the vibrations were inspected (they can be found following this link). As predicted and showed here below on an extract of the output file, the frequency calculation resulted in one imaginary frequency of magnitude -818 cm-1, meaning that job was successfully completed.
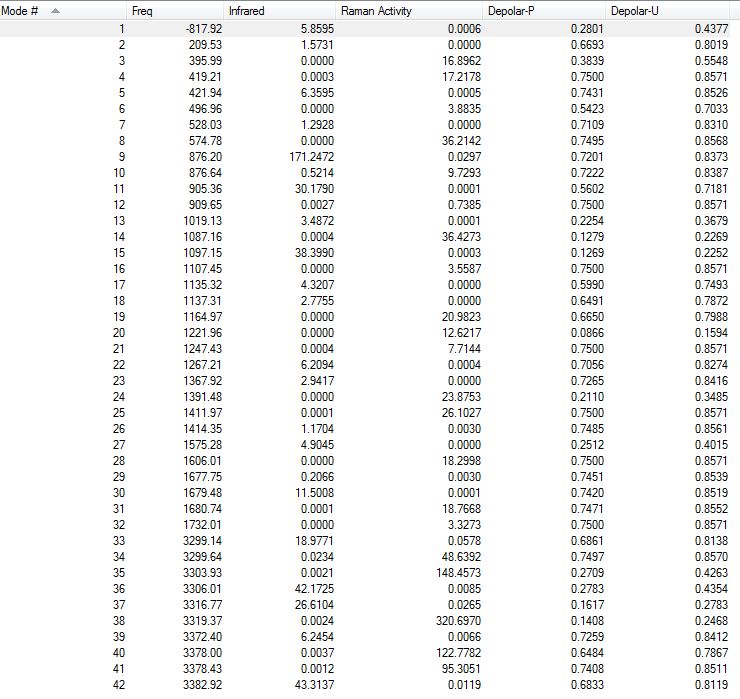{kind=link}
Low frequencies --- -817.9217 -2.6549 -0.0006 0.0004 0.0005 3.7756 Low frequencies --- 4.5940 209.5310 395.9867
This vibration, shown on the animation here below, correponds to the Cope rearrangement.

Optimisation by Freezing the Coordinates
The 'chair' structure was then optimised using a second method, as the first one often fails and/or leads to misleading results especially when the guess transition structure is far from the exact one. This time the transition structure was generated by freezing the reaction coordinate (using Opt=ModRedundant) and minimizing the rest of the molecule. After the molecule had fully relaxed, the reaction coordinate was unfrozened and the transition state was further optimised. Differentiating along the reaction coordinate was then sufficient to give a good guess for the initial force constant matrix, without having the need to compute the whole Hessian, and hence saving a considerable amount of time[4].
| Molecule | 
|
| Links to output files | log/ chk |
| Calculation Type | FOPT |
| Calculation Method | RHF |
| Basis set | 3-21G |
| Final Energy (a.u.) | -231.61465223 |
| RMS Gradient Norm (a.u.) | 0.00395651 |
| Dipole Moment (Debye) | 0.0044 |
| Point Group | C1 |
| Time of calculation (s) | 25.0 |
The guess structure of the 'chair' transition state was copied and pasted in GaussView. The terminal carbons from the allyl fragments which form/break a bond during the rearrangment were frozen. This was done by selecting 'Bond' instead of 'Unidentified' and 'Freeze Coordinate' instead of 'Add' in the Redundant Coordinate Editor. The transition structure was then optimised with the option Opt=ModRedundant.
The RMS gradient was found to be close to zero (0.00395651 a.u.), meaning that the molecule had been successfully optimised. This was further confirmed by looking at the convergence of the forces and displacements in the output files, as shown here below.
Item Value Threshold Converged? Maximum Force 0.000060 0.000450 YES RMS Force 0.000014 0.000300 YES Maximum Displacement 0.001306 0.001800 YES RMS Displacement 0.000383 0.001200 YES Predicted change in Energy=-7.406912D-08 Optimization completed. -- Stationary point found.
As shown by GaussView's snapshot (in the summary table), the optimised structure was similar to the transition state previously optimised using Hessian's method, except that the bond forming/breaking distances were fixed to 2.2 Å.
| Molecule | 
|
| Links to output files | log/ chk |
| Calculation Type | FTS |
| Calculation Method | RHF |
| Basis set | 3-21G |
| Final Energy (a.u.) | -231.61932219 |
| RMS Gradient Norm (a.u.) | 0.00005611 |
| Dipole Moment (Debye) | 0.0006 |
| Point Group | C1 |
| Point Group (after symmetrisation) | C2h |
| Time of calculation (s) | 22.0 |
Bonds distances of the previous structure were optimised by selecting the frozen bonds and selecting 'Bond' instead of 'Unidentified' and 'Derivative' instead of 'Add', in the Redundant Coordinate Editor. A transition state optimisation was then carried out, but the force constants were not calculated as it was previously done. Instead, a modified normal guess Hessian was used, so that it would include information on the two coordinates along which we would be differentiating.
The RMS gradient was found to be close to zero (0.00005611 a.u.), meaning that the molecule had been successfully optimised. This was further confirmed by looking at the convergence of the forces and displacements in the output files, as shown here below.
Item Value Threshold Converged? Maximum Force 0.000158 0.000450 YES RMS Force 0.000033 0.000300 YES Maximum Displacement 0.001644 0.001800 YES RMS Displacement 0.000461 0.001200 YES Predicted change in Energy=-2.404842D-07 Optimization completed. -- Stationary point found.
check the bond forming/bond breaking bond lengths and compare the structure to the one you optimized in section (b)
| Reactant (anti2) | Chair TS (Hessian) | Chair TS (Freezing Coordinates) | |
|---|---|---|---|
| Structure | 
|

| |
| C1-C2 | 1.31613 | 1.38931 | 1.38931 |
| C2-C3 | 1.50891 | 1.38920 | 1.38934 |
| C3-C4 | 1.55275 | 2.02069 | 2.01994 |
| C4-C5 | 1.50891 | 1.38923 | 1.38934 |
| C5-C6 | 1.31613 | 1.38937 | 1.38931 |
| C6-C1 | - | 2.02012 | 2.02065 |
Optimisation of the Boat Transition State
Using the QST2 Method
The 'boat' transition structure was then optimised, using the QST2 method. In this method, the reactants and products for a reaction need to be specified so that the calculation interpolates between the two structures to find the transition state located between them[4].
To do so, the optimised Ci anti 2 1,5-hexadiene reactant molecule, was duplicated, oriented and renumbered as shown here below, so that both the reactant and the product were numbered in the same way.
| Reactant | Product |
|---|---|
 |

|
A QST2 calculation was attempted, but the job failed: as shown by a snapshot of the checkpoint file, the obtained structure looked more like a dissociated chair transition structure (as illustrated by the relative bond distances: for example C1-C4 measures 2.02048 while it measures 2.01994 in the chair transition state)rather than a boat transition state.

This is because this calculation resulted in a linear interpolation between the two structures: it simply translated the top allyl fragment without considering the possibility of a rotation around the central bonds[4].
| C1-C2 | 1.38928 |
| C2-C3 | 1.38924 |
| C4-C5 | 1.38930 |
| C5-C6 | 1.38927 |
| C1-C4 | 2.02048 |
| C3-C6 | 2.02054 |
Revisited QST2 Method
The reactant and product geometries were modified, so that they would look closer the 'boat' transition structure[4]. To do so, the central C-C-C-C dihedral angle was set to 0o and the inside C-C-C angle was set to 100o. This gave the following geometries for the reactant and product:
| Reactant | Product |
|---|---|
 |

|
| Molecule | 
|
| Links to output files | log/ chk |
| Calculation Type | FREQ |
| Calculation Method | RHF |
| Basis set | 3-21G |
| Final Energy (a.u.) | -231.60280170 |
| RMS Gradient Norm (a.u.) | 0.00008819 |
| Dipole Moment (Debye) | 0.1578 |
| Point Group | Cs |
| Point Group (after symmetrisation) | C2h |
| Time of calculation (s) | 6.0 |
A QST2 calculation was set up again, and, this time, the job successfully converged to the boat transition structure. The log file was examined to ensure that all the forces and displacements had converged:
The RMS gradient was found to be close to zero (0.00008819 a.u.), meaning that the molecule had been successfully optimised. This was further confirmed by looking at the convergence of the forces and displacements in the output files, as shown here below.
Item Value Threshold Converged? Maximum Force 0.000093 0.000450 YES RMS Force 0.000034 0.000300 YES Maximum Displacement 0.001171 0.001800 YES RMS Displacement 0.000374 0.001200 YES Predicted change in Energy=-5.705714D-07 Optimization completed. -- Stationary point found.
As shown in the summary table, the obtained structure looked like a 'boat' transition state, and symmetrisation of the structure gave the C2v point group, as expected.
All the vibrations were inspected (they can be found following this link). As predicted and showed here below on an extract of the output file, the frequency calculation resulted in one imaginary frequency of magnitude -840 cm-1, meaning that job was successfully completed.
{kind=link}
Low frequencies --- -839.7702 -5.7517 -0.0022 -0.0011 -0.0002 2.7352 Low frequencies --- 2.8938 155.2451 381.9359
An animation of the correspoding vibration is shown here below:

Optimising the Boat Transition State via the QST3 Method
Although the QST2 method, has some advantages, for instance, it is fully automated, it can often fail if the reactants and/or products are not close to the transition structure[4]. As a result a more reliable method, called the QST3 method, which allows you to input the geometry of a guess transition structure[4], has been used for the next calculations.
As previously, the guess transition structure was renumbered, as followed, so that its numbering would match the ones of the reactant and product.

| Molecule | 
|
| Links to output files | log/ chk |
| Calculation Type | FREQ |
| Calculation Method | RHF |
| Basis set | 3-21G |
| Final Energy (a.u.) | -231.60280240 |
| RMS Gradient Norm(a.u.) | 0.00001888 |
| Dipole Moment(Debye) | 0.1583 |
| Point Group | C1 |
| Point Group (after symmetrisation) | C2v |
| Time of calculation (s) | 9.0 |
The RMS gradient was found to be close to zero (0.00001888 a.u.), meaning that the molecule had been successfully optimised. This was further confirmed by looking at the convergence of the forces and displacements in the output files, as shown here below.
Item Value Threshold Converged? Maximum Force 0.000036 0.000450 YES RMS Force 0.000010 0.000300 YES Maximum Displacement 0.001264 0.001800 YES RMS Displacement 0.000381 0.001200 YES Predicted change in Energy=-9.148480D-08 Optimization completed. -- Stationary point found.
All vibrations were inspected (they can be found following this link) and examination of the log file confirmed the presence of one imaginary frequency of magnitude -840 cm-1, as shown here below.
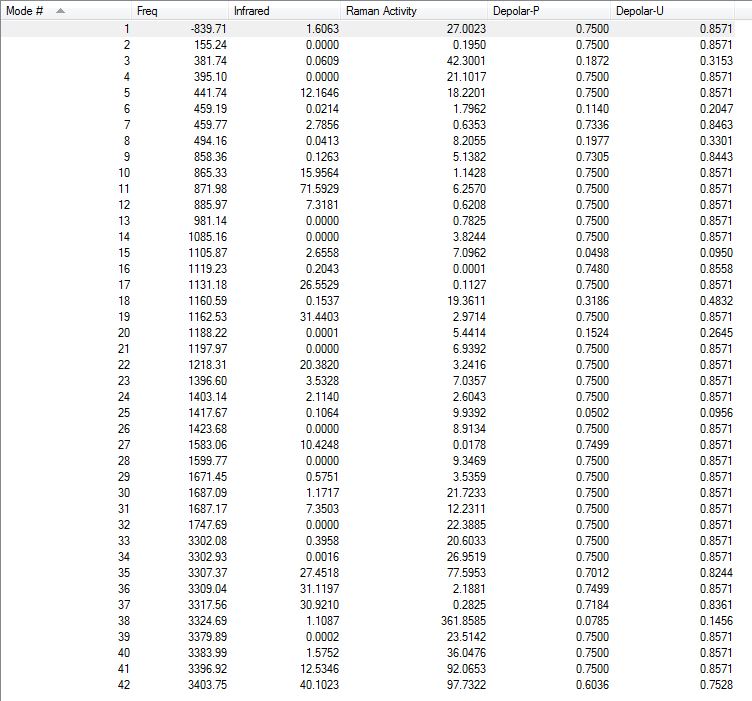{kind=link}
Low frequencies --- -839.7050 -4.0403 -0.0011 -0.0007 -0.0005 1.4918 Low frequencies --- 3.1183 155.2438 381.7431
| QST2 Method | QST3 Method | |
|---|---|---|
| Structure |  |

|
| C1-C2 | 86.989 | 1.38143 |
| C2-C3 | 1.38128 | 1.38136 |
| C3-C4 | 2.14118 | 2.14050 |
| C4-C5 | 1.38128 | 1.38140 |
| C5-C6 | 1.38140 | 1.38144 |
| C6-C1 | 2.14040 | 2.14042
|
Intrinsic Reaction Coordinates
The IRC or Intrinsic Reaction Coordinate method was then used to predict from which conformer of 1,5-hexadiene, the chair transition structure was originated. Indeed, the IRC can predict, from the transitions structures, which conformer the reaction paths will lead to. It follows the minimum energy path from a transition structure, by taking small geometry steps in the direction where the slope of the potential energy surface is steepest, until it reaches a local minimum [4].
| Molecule |  
|
| Links to output files | log/ chk |
| Calculation Type | IRC |
| Calculation Method | - |
| Basis set | 3-21G |
| Final Energy (a.u.) | -231.69157203 |
| RMS Gradient Norm (a.u.) | 0.00014489 |
| Dipole Moment (Debye) | 0.3622 |
| Point Group | C1 |
| Time of calculation | 5 min 25.0 s |
The IRC method was carried out on the optimised 'chair' transition structure. This was done by selecting 'IRC' under the Job Type tab, computing the reaction coordinate in the forward direction (as the reaction coordinate was symmetrical) and calculating the force constants at every step along the IRC. The IRC was first runned with a total number of 50 points. Calculations stopped at the last step (set 49). When running the IRC a second time, with a greater number of points (70), calculations also stopped at step 49, indicating at we had reached a local minimum on the potential energy surface.
A normal minimisation calculation was therefore runned on this point: the structure was first optimised using the Hartree Fock method with a 3-21G basis set and then further optimised using the B3LYP/6-31G(d) level of theory. This resulted in the minimum conformer gauche 2, as illustrated by the shape and final energy.

| ||
| Links to output files | log/ chk | log/ chk |
| Calculation Type | FOPT | FOPT |
| Calculation Method | RHF | RB3LYP |
| Basis set | 3-21G | 6-31G(d) |
| Final Energy (a.u.) | -231.69166702 | -234.61068493 |
| RMS Gradient Norm (a.u.) | 0.00000482 | 0.00003791 |
| Dipole Moment (Debye) | 0.3806 | 0.4392 |
| Point Group | C1 | C1 |
| Time of calculation (s) | 31.0 | 1 min 10.0 s |
Both RMS gradients were found to be close to zero (0.00000482 & 0.00003791 a.u.), meaning that structures were successfully optimised, which was further confirmed by looking at the convergence of the forces and displacements in the output files, as shown here below.
Item Value Threshold Converged? Maximum Force 0.000010 0.000450 YES RMS Force 0.000003 0.000300 YES Maximum Displacement 0.000291 0.001800 YES RMS Displacement 0.000087 0.001200 YES Predicted change in Energy=-2.429908D-09 Optimization completed. -- Stationary point found.
Item Value Threshold Converged? Maximum Force 0.000069 0.000450 YES RMS Force 0.000021 0.000300 YES Maximum Displacement 0.001723 0.001800 YES RMS Displacement 0.000512 0.001200 YES Predicted change in Energy=-7.387487D-08 Optimization completed. -- Stationary point found.
Computing Activation Energies
To calculate the activation energies for the reaction via the chair and boat transition structures, both transition structures were reoptimised using the B3LYP/6-31G(d) level of theory.
| Chair | Boat | |
|---|---|---|
| Molecule |  |

|
| Link to output file | log | log |
| Calculation Type | FREQ | |
| Calculation Method | RB3LYP | |
| Basis set | 6-31G(d) | |
| Final Energy (a.u.) | -234.55698303 | -234.54309307 |
| RMS Gradient Norm (a.u.) | 0.00001194 | 0.00000444 |
| Dipole Moment (Debye) | 0.4392 | 0.0613 |
| Point Group | C1 | C1 |
| Point Group (after symmetrisation) | C2h | C2v |
| Time of calculation | 5 min 43.8 s | 5 min 36.5 s |
Both RMS gradients were found to be close to zero (0.00001194 & 0.00000444 a.u.), meaning that structures were successfully optimised, which was further confirmed by looking at the convergence of the forces and displacements in the output files, as shown here below.
Item Value Threshold Converged? Maximum Force 0.000023 0.000450 YES RMS Force 0.000005 0.000300 YES Maximum Displacement 0.000075 0.001800 YES RMS Displacement 0.000025 0.001200 YES Predicted change in Energy=-3.724413D-09 Optimization completed. -- Stationary point found.
Item Value Threshold Converged? Maximum Force 0.000009 0.000450 YES RMS Force 0.000003 0.000300 YES Maximum Displacement 0.000169 0.001800 YES RMS Displacement 0.000054 0.001200 YES Predicted change in Energy=-3.423638D-09 Optimization completed. -- Stationary point found.
Summary of energies (in hartree)
| HF/3-21G | B3LYP/6-31G* | |||||
|---|---|---|---|---|---|---|
| Electronic energy | Sum of electronic and zero-point energies | Sum of electronic and thermal energies | Electronic energy | Sum of electronic and zero-point energies | Sum of electronic and thermal energies | |
| at 0 K | at 298.15 K | at 0 K | at 298.15 K | |||
| Chair TS | -231.619322 | -231.466701 | -231.461342 | -234.556983 | -234.414929 | -234.409009 |
| Boat TS | -231.602802 | -231.450930 | -231.445300 | -234.543093 | -234.402342 | -234.396008 |
| Reactant (anti2) | -231.692535 | -231.539539 | -231.532565 | -234.611712 | -234.469182 | -234.461850 |
*1 hartree = 627.509 kcal/mol
Summary of activation energies (in kcal/mol)
| HF/3-21G | HF/3-21G | B3LYP/6-31G* | B3LYP/6-31G* | Expt. | |
| at 0 K | at 298.15 K | at 0 K | at 298.15 K | at 0 K | |
| ΔE (Chair) | 45.71 | 44.69 | 34.04 | 33.16 | 33.5 ± 0.5 |
| ΔE (Boat) | 55.60 | 54.76 | 41.94 | 41.32 | 44.7 ± 2.0 |
The Diels Alder Cycloaddition
Part 1
Optimisation of the Reactants
Optimisation Cis-Butadiene
A molecule of cis-butadiene was created in Gaussview and optimised using the semi-empirical AM1 molecular orbtital method. It was then reoptimised using higher levels of theory: HF/3-21G followed B3LYP/6-31G(d). All RMS gradients were found to be close to zero (0.0011857, 0.0011857 & 0.00003238 a.u.), meaning that structures were successfully optimised, which was further confirmed by looking at the convergence of the forces and displacements in the output files, as shown here below.
| 1st Optimisation | 2nd Optimisation | 3rd Optimisation | |
|---|---|---|---|
| Molecule |  | ||
| Links to output files | log/ chk | log/ chk | log/ chk |
| Calculation Type | FOPT | FOPT | FOPT |
| Calculation Method | RAM1 | RHF | RB3LYP |
| Basis set | ZDO | 3-21G | 6-31G(d) |
| Final Energy (a.u.) | 0.04879734 | -154.05394305 | -155.98594961 |
| RMS Gradient Norm (a.u.) | 0.00008900 | 0.0011857 | 0.00003238 |
| Dipole Moment (Debye) | 0.0414 | 0.0338 | 0.0852 |
| Point Group | C1 | ||
| Point Group (after symmetrisation) | C2v | ||
| Time of calculation (s) | 35.0 | 23.0 | 40.0 |
Item Value Threshold Converged?
Maximum Force 0.000159 0.000450 YES
RMS Force 0.000051 0.000300 YES
Maximum Displacement 0.000661 0.001800 YES
RMS Displacement 0.000254 0.001200 YES
Predicted change in Energy=-1.540760D-07
Optimization completed.
-- Stationary point found.
Item Value Threshold Converged?
Maximum Force 0.000239 0.000450 YES
RMS Force 0.000078 0.000300 YES
Maximum Displacement 0.000995 0.001800 YES
RMS Displacement 0.000343 0.001200 YES
Predicted change in Energy=-2.308366D-07
Optimization completed.
-- Stationary point found.
Item Value Threshold Converged?
Maximum Force 0.000073 0.000450 YES
RMS Force 0.000023 0.000300 YES
Maximum Displacement 0.000266 0.001800 YES
RMS Displacement 0.000132 0.001200 YES
Predicted change in Energy=-2.716651D-08
Optimization completed.
-- Stationary point found.
The HOMO and LUMO were visualised using the checkpoint file :
| Calculation Method | Energies | MO |
|---|---|---|
AM1 |
-0.34382 | 
|
HF |
-0.32529 | |
| B3LYP | -0.22736 |
| Calculation Method | Energies | MO |
|---|---|---|
AM1 |
0.01709 | 
|
HF |
0.12353 | |
| B3LYP | -0.03013 |
BOTH ANTISYMMETRIC
Optimisation of Ethylene
| Molecule | 
|
| Links to output files | log/ chk |
| Calculation Type | FOPT |
| Calculation Method | RB3LYP |
| Basis set | 6-31G(d) |
| Final Energy (a.u.) | -78.58745746 |
| RMS Gradient Norm (a.u.) | 0.00014422 |
| Dipole Moment (Debye) | 0.0000 |
| Point Group | C1 |
| Point Group (after symmetrisation) | D2h |
| Time of calculation | 7.0 s |
A molecule of ethlyne was created in Gaussview and optimised using a B3LYP method and a 6-31G(d) basis set. Its RMS gradient was found to be close to zero (0.00014422 a.u.), meaning that the structure had been successfully optimised. This was further confirmed by looking at the convergence of the forces and displacements in the output files, as shown here below.
Item Value Threshold Converged? Maximum Force 0.000230 0.000450 YES RMS Force 0.000136 0.000300 YES Maximum Displacement 0.001375 0.001800 YES RMS Displacement 0.001006 0.001200 YES Predicted change in Energy=-8.331632D-07 Optimization completed. -- Stationary point found.
| HOMO | LUMO | |
|---|---|---|
| Snapshot |  |

|
| Energy | -0.26667 | +0.01881 |
| Cis-Butadiene | HOMO | LUMO |
| Ethylene | LUMO | HOMO |
| ΔE (HOMO-LUMO) | 0.24617 | 0.23654 |
LUMO of cis-butadiene with HOMO of ethylene reacting
Optimisation of the Product: Cyclohexene
| Molecule | 
|
| Links to output files | log/ chk |
| Calculation Type | FOPT |
| Calculation Method | RB3LYP |
| Basis set | 6-31G(d) |
| Final Energy (a.u.) | -234.63289313 |
| RMS Gradient Norm (a.u.) | 0.00007877 |
| Dipole Moment (Debye) | 0.3498 |
| Point Group | C1 |
| Point Group (after symmetrisation) | C2v |
| Time of calculation | 1 min 31.0 s |
A molecule of cyclohexene was created in GaussView and optimised using a B3LYP method and 6-31G(d)basis set. The RMS gradient was found to be close to zero (0.00007877 a.u.), meaning that the structure had been successfully optimised. This was further confirmed by looking at the convergence of the forces and displacements in the output files, as shown here below.
Item Value Threshold Converged? Maximum Force 0.000207 0.000450 YES RMS Force 0.000044 0.000300 YES Maximum Displacement 0.000975 0.001800 YES RMS Displacement 0.000252 0.001200 YES Predicted change in Energy=-2.989012D-07 Optimization completed. -- Stationary point found.
Optimisation of the Transition State
The transition state was optmised using two different methods.
Using the Hessian Method
The previously optimised cis-butadiene and ethylene fragments were oriented such that they would like the required transition state, seperating terminal ends by approximately 2.2 Å. The transition state was then optimised using the Hartree Fock method and 3-21G basis set. To do so, the 'Optimisation' command was set to a TS (Berny)', force constants were only calculated once, and the additional keywords Opt=NoEigen were included. The obtained structure was then further optimised using a higher level of theory: B3LYP/6-31G(d).
| 1st Optimisation | 2nd Optimisation | |
|---|---|---|
| Molecule |  
| |
| Links to output files | log | log/ chk |
| Calculation Type | FREQ | FREQ |
| Calculation Method | RHF | RB3LYP |
| Basis set | 3-21G | 6-31G(d) |
| Final Energy (a.u.) | -231.60320857 | -234.54389653 |
| RMS Gradient Norm (a.u.) | 0.00000342 | 0.00001338 |
| Dipole Moment (Debye) | 0.5757 | 0.3943 |
| Point Group | C1 | |
| Point Group (after symmetrisation) | Cs | |
| Time of calculation (s) | 21.6 | 40.0 |
Both RMS gradients were found to be close to zero (0.00000342 & 0.00001338 a.u.), meaning that structures were successfully optimised, which was further confirmed by looking at the convergence of the forces and displacements in the output files, as shown here below.
Item Value Threshold Converged? Maximum Force 0.000014 0.000450 YES RMS Force 0.000002 0.000300 YES Maximum Displacement 0.000030 0.001800 YES RMS Displacement 0.000009 0.001200 YES Predicted change in Energy=-3.866693D-10 Optimization completed. -- Stationary point found.
Item Value Threshold Converged? Maximum Force 0.000030 0.000450 YES RMS Force 0.000006 0.000300 YES Maximum Displacement 0.000625 0.001800 YES RMS Displacement 0.000172 0.001200 YES Predicted change in Energy=-2.365409D-08 Optimization completed. -- Stationary point found.
Using the QST3 Method
The guessed transition structure was then optimised using the QST3 method with the B3LYP/6-31G(d)level of theory. To do so, both reactants and the product were used in the input file.
| Molecule |  
|
| Links to output files | log/ chk |
| Calculation Type | FREQ |
| Calculation Method | RB3LYP |
| Basis set | 6-31G(d) |
| Final Energy (a.u.) | -234.54389655 |
| RMS Gradient Norm (a.u.) | 0.00001904 |
| Dipole Moment (Debye) | 0.3943 |
| Point Group | C1 |
| Point Group (after symmetrisation) | Cs |
| Time of calculation | 1 min 22.0 s |
The RMS gradient was found to be close to zero (0.00001904 a.u.), meaning that the structure had been successfully optimised, which was further confirmed by looking at the convergence of the forces and displacements in the output files, as shown here below.
Item Value Threshold Converged? Maximum Force 0.000016 0.000450 YES RMS Force 0.000006 0.000300 YES Maximum Displacement 0.001238 0.001800 YES RMS Displacement 0.000245 0.001200 YES Predicted change in Energy=-8.219250D-09 Optimization completed. -- Stationary point found.
All the vibrations were inspected (they can be found following this link). As expected for a transition structure, the frequency calculation resulted in one imaginary frequency, of magnitude -526 cm-1, as shown here below, meaning that job was successfully completed.
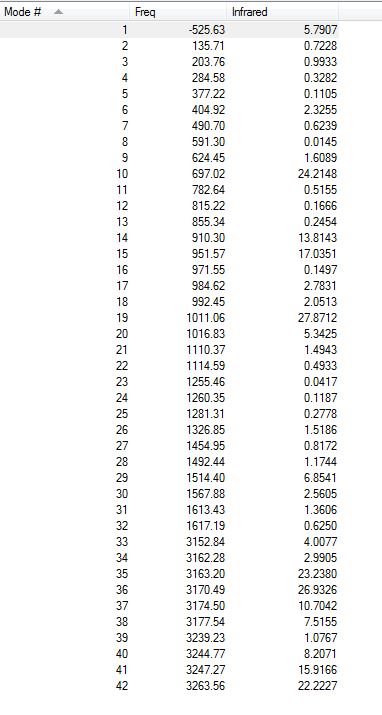{kind=link}
Low frequencies --- -525.6297 -6.7129 0.0005 0.0006 0.0010 10.1114 Low frequencies --- 19.8223 135.7697 203.7765
An animation of the corresponding vibration is shown here below:

Bond forming/breaking distances of the optimised structures of the guessed transition state were tabulated. As shown here below, those distances are relatively similar using both methods of optimisation.
| Bonds | Reactants | Transition State (Hessian Method) | Transition State (QST3 Method) | |
|---|---|---|---|---|
 |
C1-C2/C6-C7 | 1.33985 | 1.38303/1.38306 | 1.38310 |
| C1-C6 | 1.47144 | 1.40717 | 1.40712 | |
| C11-C12 | 1.33071 | 1.38598 | ||
| C2-C11/C7-C12 | - | 2.27244/2.27234 | 2.27175/2.27171 |
| HOMO | LUMO |
|---|---|
 |

|
 |

|
 |

|
As illustrated by the shape of the HOMO; HOMO of ethylene reacting with LUMO of cis-butadiene
Part 2 : Study the Regioselectivity of the Diels Alder Reaction
The regioselectivity of Diels Alder's reaction was investigated by looking at the reaction of cyclohexa-1,3-diene with maleic anhydride. This reaction is supposedly under kinetic control and should primarily give the endo adduct.
Optimisation of the Reactants and Products
Both reactants and products were optimised using a B3LYP method and a 6-31G(d) basis set.
| Cyclohexa-1,3-diene | Maleic Anhydride | Exo Adduct | Endo Adduct | |
|---|---|---|---|---|
| Molecule |  |
 |
 |

|
| Links to output files | log/ chk | log/ chk | log/ chk | log/ chk |
| FOPT | ||||
| RB3LYP | ||||
| 6-31G(d) | ||||
| Final Energy (a.u.) | -233.41588484 | -379.28954441 | -612.75578548 | -612.75829019 |
| RMS Gradient Norm (a.u.) | 0.00007566 | 0.00010583 | 0.00000675 | 0.00001632 |
| Dipole Moment (Debye) | 0.5484 | 4.0718 | 4.7598 | 5.0201 |
| Point Group | C1 | |||
| Point Group (after symmetrisation) | C2v | C2v | Cs | Cs |
| Time of calculation | 1 min 16.0 | 1 min 3.0 | 21 min 42.0 | 25 min 43.0 |
RMS gradients were found to be close to zero (0.00007566, 0.00010583, 0.00000675 & 0.00001632 a.u.), meaning that the structures were successfully optimised. This was further confirmed by looking at the convergence of the forces and displacements in the output files, as shown here below.
Item Value Threshold Converged? Maximum Force 0.000275 0.000450 YES RMS Force 0.000038 0.000300 YES Maximum Displacement 0.000532 0.001800 YES RMS Displacement 0.000141 0.001200 YES Predicted change in Energy=-1.439580D-07 Optimization completed. -- Stationary point found.
Item Value Threshold Converged? Maximum Force 0.000216 0.000450 YES RMS Force 0.000071 0.000300 YES Maximum Displacement 0.000692 0.001800 YES RMS Displacement 0.000250 0.001200 YES Predicted change in Energy=-2.569410D-07 Optimization completed. -- Stationary point found.
Item Value Threshold Converged? Maximum Force 0.000019 0.000450 YES RMS Force 0.000004 0.000300 YES Maximum Displacement 0.000814 0.001800 YES RMS Displacement 0.000143 0.001200 YES Predicted change in Energy=-1.243639D-08 Optimization completed. -- Stationary point found.
Item Value Threshold Converged? Maximum Force 0.000040 0.000450 YES RMS Force 0.000007 0.000300 YES Maximum Displacement 0.001553 0.001800 YES RMS Displacement 0.000230 0.001200 YES Predicted change in Energy=-2.658569D-08 Optimization completed. -- Stationary point found.
| HOMO of Cyclohexa-1,3-diene | LUMO of Maleic Anhydride | |||
|---|---|---|---|---|
| MOs |  |
 |
 |

|
| Energies [B3LYP/6-31G(d)] | -0.20102 | -0.11709 | ||
| ΔE(HOMO-LUMO) | 0.31811 | |||
Optimisation of the Transition States
Structures of both transition states - the one corresponding to the exo adduct and the one corresponding to the endo adduct - were guessed and then optimised using the QST3 method. To do so, structures of the reactants, transition states and products, were added to the input files, renumbered and oriented properly. The Job title was changed to 'Opt+Freq', the 'Optimisation' command was set to a 'QTS3' and force constants were only calculated at every step (to maximise changes of finding of the transition state). The transition states were optimised using a B3LYP method and a 6-31G(d) basis set.
Optimisation of the Exo Transition State
| Molecule | 
|
| Links to output files | log/ chk |
| Calculation Type | FREQ |
| Calculation Method | RB3LYP |
| Basis set | 6-31G(d) |
| Final Energy (a.u.) | -612.67931096 |
| RMS Gradient Norm (a.u.) | 0.00000288 |
| Dipole Moment (Debye) | 5.5501 |
| Point Group | C1 |
| Point Group (after symmetrisation) | Cs |
| Time of calculation | 26 min 23.6 s |
The RMS gradient was found to be close to zero (0.00000288 a.u.), meaning that the structure was successfully optimised. This was further confirmed by looking at the convergence of the forces and displacements in the output files, as shown here below.
Item Value Threshold Converged? Maximum Force 0.000004 0.000450 YES RMS Force 0.000001 0.000300 YES Maximum Displacement 0.000434 0.001800 YES RMS Displacement 0.000126 0.001200 YES Predicted change in Energy=-3.909007D-09 Optimization completed. -- Stationary point found.
All the vibrations were inspected (they can be found following this link). As expected for a transition structure, the frequency calculation resulted in one imaginary frequency, of magnitude -448 cm-1, as shown here below, meaning that job was successfully completed.
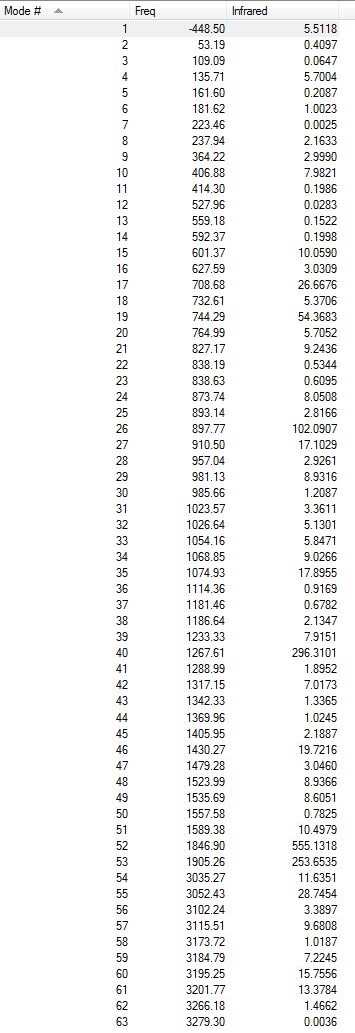{kind=link}
Low frequencies --- -448.4969 -13.9288 -11.7631 0.0008 0.0009 0.0010 Low frequencies --- 3.0620 53.3059 109.0927
An animation of the corresponding vibration is shown here below:

Optimisation of Endo TS
| Molecule | 
|
| Links to output files | log/ chk |
| Calculation Type | FOPT |
| Calculation Method | RB3LYP |
| Basis set | 6-31G(d) |
| Final Energy (a.u.) | -612.68339677 |
| RMS Gradient Norm (a.u.) | 0.00007877 |
| Dipole Moment (Debye) | 0.3498 |
| Point Group | C1 |
| Point Group | Cs |
| Time of calculation | 26 min 6.7 s |
The RMS gradient was found to be close to zero (0.00007877 a.u.), meaning that the structure was successfully optimised. This was further confirmed by looking at the convergence of the forces and displacements in the output files, as shown here below.
Item Value Threshold Converged? Maximum Force 0.000003 0.000450 YES RMS Force 0.000000 0.000300 YES Maximum Displacement 0.000359 0.001800 YES RMS Displacement 0.000066 0.001200 YES Predicted change in Energy=-9.966983D-10 Optimization completed. -- Stationary point found.
All the vibrations were inspected (they can be found following this link). As expected for a transition structure, the frequency calculation resulted in one imaginary frequency, of magnitude -447 cm-1, as shown here below, meaning that job was successfully completed.
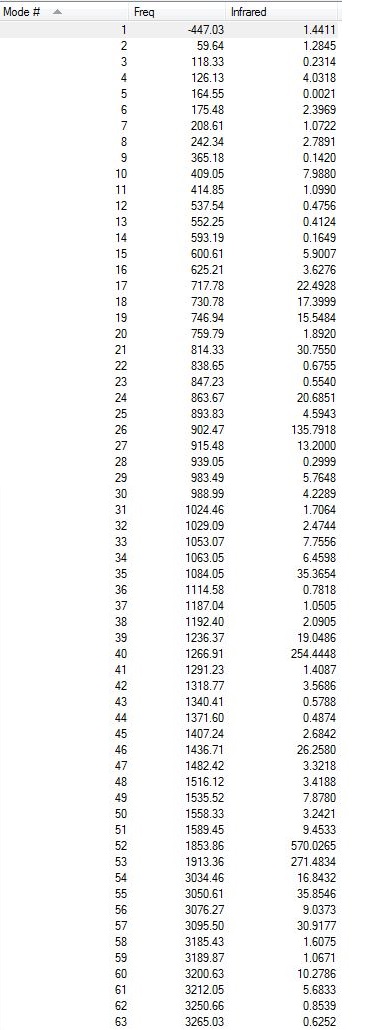{kind=link}
Low frequencies --- -447.0280 -14.2619 -0.0004 0.0005 0.0005 4.5194 Low frequencies --- 11.2870 59.6502 118.3544
An animation of the corresponding vibration is shown here below:

Discussion on the relative stabilities of the Transition States
| Exo Transition State | Endo Transition State | |
|---|---|---|
| Structure |  |

|
| C1-C4 (Double Bond forming) | 1.40345 | 1.40307 |
| C1-C2/C4-C3 (Double Bonds breaking) | 1.39140 / 1.39142 | 1.39124 / 1.39125 |
| C2-C12/C3-C9 | 1.51453 | 1.51508 |
| C12-C9 | 1.55839 | 1.55823 |
| C3-C17/C2-C16 (Bond between cyclohexa-1,3-diene and maleic anhydride forming) | 2.29046 / 2.29068 | 2.26841/ 2.26829 |
| C16-C17 (Double Bond breaking) | 1.39790 | 1.39397 |
| Through space interaction | C9-C18/C12-C15: 3.02793 / 3.02788 | C1-C18/C4-C15: 2.99007 / 2.99016 |
| Exo Transition State | Endo Transition State |
|---|---|
| -612.67931096 | -612.68339677 |
endo=lower in energy, more stable => smaller activation energy => endo= favoured More steric repulsion in the endo TS (smaller distances) but secondary overlap interaction !
| Exo Transition State | Endo Transition State | |||
|---|---|---|---|---|
| HOMO |  |
 |
 |

|
| LUMO |  |
 |
 |

|
References
- ↑ 1.0 1.1 1.2 1.3 1.4 1.5 1.6 P. Hunt, Inorganic Module: Bonding and Molecular Orbitals in main group compounds, 3rd Year Computational Lab, Imperial College 2013
- ↑ 2.0 2.1 2.2 2.3 2.4 H. Rzepa, Conformational Analysis, 2rd Year Organic Chemistry, Imperial College 2012
- ↑ 3.0 3.1 3.2 3.3 3.4 3.5 B. W. Gung, Z. Zhu, and R. A. Fouch, J. Am. Chem. Soc., 1995, 117, 1783-1788
- ↑ 4.00 4.01 4.02 4.03 4.04 4.05 4.06 4.07 4.08 4.09 M. Bearpark, Physical Module: Transition States and Reactivity, 3rd Year Computational Lab, Imperial College 2013