
Rep:Mod:phys3TLK2740
Diels-Alder cycloaddition reaction
The Diels-Alder reaction is the most famous example of a cycloaddition reaction. It is a [4,2] cycloaddition reaction and it is widely used in synthesis. In this exercise two Diels Alder reactions were examined. The first was the Diels-Alder reaction of cis butadiene with ethane which is the most simple Diels-Alder reaction possible. The second, more complicated one was the Diels-Alder cycloaddition of maleic anhydride to cyclohexa-1,3-diene.
Optimisation of cis butadiene


Cis-butadiene, one of the reactants for the first Diels-Alder reaction, was drawn and optimised using the AM1 semi-empirical molecular orbital method. The results are given below:
Energy: 0.04878534 a.u.
Point group: C2
The prediction of the structure of the HOMO and LUMO of the molecule by this method is shown in the figures on the right and on the left respectively.
Based on this prediction the HOMO of cis-butadiene is antisymmetric with respect to a plane passing through the middle of the molecule, while the LUMO is symmetric with respect to the plane.
Computation of the transition state geometry for the cis butadiene – ethane reaction
An assumed envelope type transition structure was drawn for the cycloaddition reaction with an initial distance of 2.2 Å kept between the two reactants. This structure was optimised using the HF/3-21G level of theory to a transition state (method 1) and a frequency calculation was obtained. In order to confirm the structure obtained, the optimisation was repeated however this time the frozen coordinate method was used (method 2). By initially freezing the bond forming/breaking distance at 2.2 Å, optimising the structure and then optimising the bond distance as well, a result similar to that of the first optimisation was obtained. The results are shown in the table below:

The transition structure obtained by the frozen coordinates method (method 2) was reoptimised using the B3LYP/6-31G* level of theory as it is mentioned in literature as one of the most accurate when calculating the energy barriers in pericyclic reactions [1].
Energy: -234.559730 a.u.
Imaginary frequency: 527.95 cm-1
Bond forming distance: 2.26772 Å
Angle of approach: 102.32°
Point group: CS
The values obtained are close to values found in literature[2][3].

(The values for what? Activation energy? João (talk) 12:35, 24 December 2014 (UTC))
The bond forming distance (2.21 Å) is longer than the typical sp3 and sp2 bond lengths (1.54 Å and 1.33 Å respectively). It is however shorter than the sum of the van der Waals radii of the two interacting carbons (1.70 + 1.70 = 3.40 Å). This means that the molecules are driven to react by their van der Waals interaction.
The animation of the imaginary frequency in each case showed that the vibrations correspond to the Diels-Alder reaction path. This proves that the transition state was detected successfully. From the animation it is evident the formation of the bonds during the reaction is synchronous (concerted).
![]()
In contrast the vibration corresponding to the lowest positive frequency obtained appears to be perpendicular to the one corresponding to the reaction path. The molecules appear to rotate in opposite directions.



The HOMO and LUMO orbitals of the determined transition state were visualised based on the results of the HF/3-21G calculation. These were chosen over the B3LYP/6-31G* calculation results due to the fact that the orbitals are more diffuse and it is easier to determine their symmetry.
The HOMO is symmetric with respect to a plane. The LUMO is also symmetric.

The HOMO of the transition state was formed by the interaction of the HOMO of ethene with the LUMO of cis-butadiene, both symmetric with respect to a plane passing through their middle (suprafacial components), or by the interaction of the LUMO of ethene and the HOMO of cis-butadiene (both antisymmetric), since the reaction can go both ways[4]. (Combining two orbitals of two fragments each anti-symmetric with respect to the vertical plane, how do you obtain a symmetric orbital? João (talk) 12:35, 24 December 2014 (UTC))
This reaction is thermally allowed (Woodward-Hoffmann rules; 4n+2 π electrons, suprafacial stereochemistry, Hückel transition state topology).
The resulting HOMO of the transition state is suprafacial.
Diels-Alder reaction stereoselectivity: cycloaddition of maleic anhydride to cyclohexa-1,3-diene
The transition structures for the formation of the exo and endo products of this reaction were located. This was done by drawing both the reactants and the respective product and performing a QST2 calculation to find the transition state lying between them.
First, cyclohexa-1,3-diene was optimised to its lowest energy conformation based on the HF/3-21G level of theory. Then the same was done to maleic anhydride. Next, the two possible products, first the exo and then the endo, were drawn and optimised. Two QST2 calculations were run, one for the exo product and one for the endo product. The resulting transition structures were reoptimised on the B3LYP/6-31G* level of theory. The results are given in table:



From the results obtained it is calculated that the exo transition state is more stable than the endo (This is not what your table says. The endo transition state has a more negative energy. João (talk) 12:35, 24 December 2014 (UTC)). This difference is found to be equal to 0.006777 a.u. (4.253 kcal/mol) in the HF/3-21G calculation and 0.003971 a.u. (2.492 kcal/mol) in the B3LYP/6-31G* calculation.
The transition state structures obtained from the B3LYP/6-31G* calculation are shown below. The exo structure is on the left and the endo on the right. All the distances are in Angstrom.
The exo transition state is more strained than the endo due to the repulsive interaction of the maleic anhydride with the -CH2CH2- part of the cyclohexa-1,3-diene. In the exo molecule the distance between them is smaller than the sum of the carbon van der Waals radii (3.40 Å), while in the endo the distance is larger so there is no interaction (Indeed, for the endo structure the CH2 groups are in the opposing side of the molecule. For a fair comparison of steric effects, shouldn't you compare between closer lying atoms? João (talk) 12:35, 24 December 2014 (UTC))
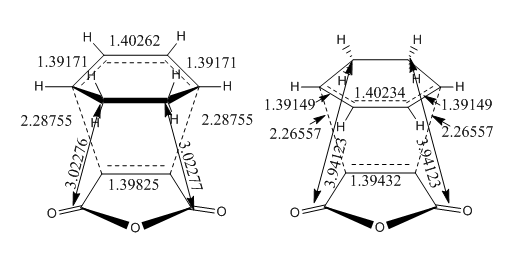
In the molecular orbital diagrams of the HOMO of the exo and the endo transition states it is observed that the electron density in the -(C=O)-O-(C=O)- part is much lower than in the rest of the molecule. This could be attributed to secondary orbital overlap.[5]There is an extra bonding interaction between the frontier orbitals of cyclohexa-1,3-diene and maleic anhydride (How does low electron density contribute to the stabilization? There could be secondary orbital effects for other orbitals lower in energy than the HOMO though João (talk) 12:35, 24 December 2014 (UTC)). The interaction does not result in the formation of bonds, however it results in decreased electron density in the -(C=O)-O-(C=O)- part of the maleic anhydride. Due to this secondary overlap the endo transition state is stabilised more than the exo transition state and should therefore have lower energy.
References
- ↑ Bachrach, S.M. (2007) Computational organic chemistry, United States of America: John Wiley & sons, p.125.
- ↑ B. Jursic and Z. Zdravkovski, "DFT study of the Diels-Alder reactions between ethylene with buta-1,3-diene and cyclopentadiene", J. Chem. Soc. Perkin Trans., 1995, 2, 1223-1226.DOI:10.1039/P29950001223
- ↑ T. C. Dinadayalane, R. Vijaya, A. Smitha, and G. Narahari Sastry, "Diels-Alder Reactivity of Butadiene and Cyclic Five-Membered Dienes ((CH)4X, X=CH2,SiH2, O, NH, PH, and S) with Ethylene: A Benchmark Study", J. Phys. Chem., 2002, 106, 1627-1633.DOI:10.1021/jp013910r
- ↑ Fleming, I. (1990) Frontier orbitals and organic reactions, 2nd edn., Great Britain: John Wiley & sons, p.88.
- ↑ Fleming, I. (1990) Frontier orbitals and organic reactions, 2nd edn., Great Britain: John Wiley & sons, p.107.