
Rep:Mod:lyjxiong
Techniques of molecular mechanics and semi-empirical molecular orbital methods for structural and spectroscopic evaluations
The Hydrogenation of Cyclopentadiene Dimer
Based on the results of experiments, molecular mechanics technique can be used to determine whether cyclodimerisation of cyclopentadiene and hydrogenation of the dimeris under is kinetically or thermodynamically controlled by comparing related energy of two possible products.
Set-up
Program:ChemBio3D Ulta v.12.0
Method: MM2 Force-field
Results
Dimerisation of cyclopentadiene
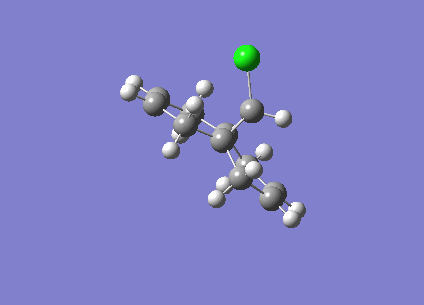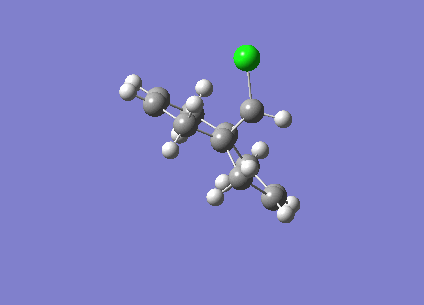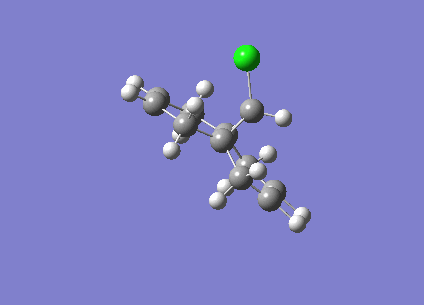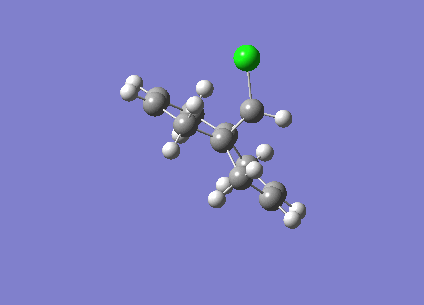
Fig1. Structures of Isomers of Cyclopentadiene Dimerises

Table1: MM2 calculated data for cyclopentadiene dimers(Units in kcal/mol)
| Dimer | MM2 Energy | Stretch | Bend | Stretch bend | Torsion | Non- 1,4 VDW | 1,4 VDW | Dipole/Dipole |
|---|---|---|---|---|---|---|---|---|
| Molecule 1 | 31.8765 | 1.2858 | 20.5796 | -0.8387 | 7.6544 | -1.4156 | 4.2334 | 0.3775 |
| Molecule 2 | 33.9975 | 1.2507 | 20.8473 | -0.8355 | 9.5122 | -1.5449 | 4.3210 | 0.4477 |
| Energy Difference(M2-M1) | 2.1210 | -0.0351 | 0.2677 | -0.0032 | 1.8578 | -0.1293 | 0.0876 | 0.0702 |
Cyclopentadiene reacts to form dicyclopentadiene undergoes a Diels-Alder π4s + π2s cycloaddition. There are two potential products formed in this reaction as shown in the figure1, however, endo-dimer has been found to the predominant product[1]. In order to determine whether the reaction is under kinetically or thermodynamically controlled, both kinetic and thermodynamical factors should be discussed.
As can be observed in the table1, the energy of endo dimer (molecule 2) is roughly 2 kcal/mol higher than that of exo dimer (molecule 1). Hence, the exo-dimer is a more stable isomer.By the consideration of result endo-dimer formed as a predominant product,the reaction can be determined to be under kinetic control.
Reactions under kinetic control are generally irreversible, the product formations rely on the stability of the transition state instead of the stability of the product. The diagram of transition states of both dimer are shown below, the transition state of endo-dimer is stabler due to more secondary orbitals overlap.So the endo-dimer forms as the product under kinetic control, this agrees with experiment results mentioned above.

The Hydrogenation of Cyclopentadiene Dimer
The predominant product (endo-dimer) from dimerisation of cyclopentadiene can undergo hydrogenation reaction to form two potential dihydro derivatives as shown in below.
Fig2. Structures of isomers of dihydro derivatives

Table2. MM2 calculated data for dihydro derivatives (units in kcal/mol)
| Dimer | MM2 Energy | Stretch | Bend | Stretch bend | Torsion | Non- 1,4 VDW | 1,4 VDW | Dipole/Dipole |
|---|---|---|---|---|---|---|---|---|
| Molecule 3 | 35.9226 | 1.2346 | 18.9429 | -0.7610 | 12.1177 | -1.4989 | 5.7284 | 0.1631 |
| Molecule 4 | 29.2475 | 1.1298 | 13.0134 | -0.5652 | 12.4121 | -1.3247 | 4.4411 | 0.1410 |
| Energy Difference (M3-M4) | 6.6751 | 0.1058 | 5.9295 | -0.1958 | -0.7056 | -0.1742 | 1.2873 | 0.0221 |
As can be observed in the table2, the molecule 4 has lower energy than that of molecule 3 by 6.6751 kcal/mol. This can be rationalised by the obvious difference of bending energy (5.9295 kcal/mol). Comparing the two possible products, the C-C double bond is in the six-member ring with a carbon bridge for molecule 3, while the C-C double bond is in the five-member ring for molecule 4. As there is a carbon bridge in the the the six-member ring, the presence of carbon double bond causes larger bending energy than the molecule with carbon double bond in five-member ring.
The molecule 4 has been determined to be the predominent product[2], this can be explained by the reason that it is easier to hydrogenate the double bond with larger bending energy to form the molecule 4. The ease of hydrogenation of double bond in this reaction can be also used to explain the reason why tetrahedro derivative is only formed after prolonged hydrogenation. Hence, this reaction is under kinetic control eventhough the thermodynamic factor also prefers the fomation of the molecule 4 because the stability of the products is not the key factor to control the reaction.
A key intermediate 9 or 10, which are atropisomers in the total synthesis of Taxol is initially synthesised with the carbonyl group pointing either up or down. Molecular Mechanics can be used to determine the stability of the isomer 9 and 10, and rationalise the fomation of atropisomers as well as the reason why the alkene reacts slowly.
Set-up
Program:ChemBio3D ulta v.12.0
Methods:MM2 force-field and MMFF94 field
Results
Stability of the isomer 9 and 10
Fig5. images of molecule 9 and 10

T3. Eenergy of the two atropisomers using an MM2 forcefield (kcal/mol)
| Molecule | MM2 Energy | Stretch | Bend | Stretch bend | Torsion | Non- 1,4 VDW | 1,4 VDW | Dipole/Dipole |
|---|---|---|---|---|---|---|---|---|
| Molecule 9 | 48.5289 | 2.7027 | 15.8871 | 0.4178 | 19.3014 | -1.3943 | 13.2862 | -1.6722 |
| Molecule 10 | 41.3808 | 2.6025 | 10.4214 | 0.2912 | 19.3724 | -2.2170 | 12.9194 | -2.0092 |
| Energy Difference(M9-M10) | 7.1481 | 0.1002 | 5.4657 | 0.1266 | -0.0710 | 0.8227 | 0.3668 | 0.3370 |
T4. Eenergy of the two atropisomers using an MMFF94 forcefield (kcal/mol)
| Molecule | MM2 Energy |
|---|---|
| Molecule 9 | 73.8697 |
| Molecule 10 | 61.1936 |
| Energy Difference(M9-M10) | 12.6761 |
As can be observed from two tables above of different methods of energy minimisation calculations, the isomer with the carbonyl group pointing down( molecule 10)is more stable by 7.1481 and 12.6761 kcal/mol respectively. Hence the molecule 10 is suppose to convert to melecule 9. However, both molecule 9 and 10 have chair conformation, which have higher energy. When an a atropisomer with higher energy converts to the one with lower energy, it needs to flip to form the chair conformation state. This not favourable because it requires extra energy to conquer the energy barrier. The energy of chair conformations are shown below.
T5. Eenergy of the two atropisomers (chair forms) using both MM2 an MMFF94 forcefields (kcal/mol)
| Molecule | MM2 Energy | MMFF94 |
|---|---|---|
| Molecule 9' | 47.2190 | 65.7603 |
| Molecule 10' | 53.0101 | 77.9633 |
| Energy Difference(M9-M10) | 5.7911 | 12.2027 |
From the table5, the both chair conformations are roughly 5 kcal/mol high in energy, and the isomer with he carbonyl group pointing down(molecule 10') is stabler in energy. This agrees with literature which demonstrates the isomer with the carbonyl group pointing down is more stable by around 4kcal/mol, as well as the result that the more stable isomers formed with 80% proportion after heating a mixture of the atropisomers.[3]
Hydrogenation Analysis
The isomer molecule 10 now is used to analyse the hydrogenation of this molecule. The picture of the product of hydrogenation of molecule10 is shown below, MM2 an MMFF94 forcefields are used to determine the stability of molecule10 and the hydrogenated molecule10.
Fig6. Image of hydrogenated molecule10

T6. Eenergy of the Molecule10 and Molecule10(Hydrogenated) using both MM2 an MMFF94 forcefields (kcal/mol)
| Molecule | MM2 Energy | MMFF94 |
|---|---|---|
| Molecule 10 | 41.3808 | 61.1936 |
| Molecule 10(Hydrogenated) | 49.8221 | 69.9432 |
| Energy Difference(M10(H)-M10) | 8.4413 | 8.7496 |
Hydrogenation of molecule10 has been demonstrated to be reacted abnormally slowly. The alkene group is next to the carbon bridge, which is expected to react quickly because of the strained environment.This can be explained by the concept of “hyperstable” olefins. Such olefins are stabilized rather than destabilized because of their location at a bridgehead and should be thermodynamically more stable than any of their positional isomers[4].Hence, hyperstable olefins should be remarkably unreactive. Moreover, the miniminsation energy shown in table 6 also agrees with this concept, where the hydrogenated molecule10 has high energy than molecule by around 8 kcal/mol.
Modelling Using Semi-empirical Molecular Orbital Theory
Regioselective Addition of Dichlorocarbene to a diene
In the previous two parts, mechanical molecular model was used to determine energy for the comparison of the stability of related molecules. However, the weakness, such as it cannot handle the interactions of orbitals has been shown.In this section, a variety of methods were used to illustrate such electronic aspects of reactivity, showing how explicit consideration of the electrons in molecules must be taken into account, and how the electrons influence bonds and derived spectroscopic properties.
Fig7, Image of molecule 12

The image of endo-prodcut from molecule 12 from literature[5]
![The image of endo-prodcut from molecule 12 from literature[5]](/images/3/30/12PPICLYJ.PNG){kind=link}
The literature[5] has reported that the molecule 12 reacts with electrophilic reagents dichlorocarbene regiospecifically on the double bond endo to the chlorine substituent, on the face of the molecule opposite the cyclopropylring. Modelling Using Semi-empirical Molecular Orbital Theory can be used to ecplain why this happens.
T7. Table of MO diagrams
| MO | Energy | MO Images |
|---|---|---|
| HOMO-1 | -0.24519 | 
|
| HOMO | -0.23588 | 
|
| LUMO | 0.01555 | 
|
| LUMO+1 | 0.02025 | 
|
| LUMO+2 | 0.03613 | 
|
From the literature[5],the exo pi-orbital (HOMO-1) is stablised by antiperiplanar overlap with the Cl-C sigma* (LUMO+2) orbital by 0.08 eV (0.931ev from calculation) relative to the endo pi-orbital (HOMO). Hence, the endo pi-orbital is more favorable to donate electrons to electrophiles. The HOMO-1 orbital is also found to been more delocalised with x-type cyclopropyl orbital, which causes smaller p-pi orbital coefficient on each of exo compared to the endo alkene carbons. Both these two factors result explains endo alkene is more reactive towards an electrophilic reagent.Hence, in particular, the molecule 12 reacts with electrophilic reagents dichlorocarbene regiospecifically on the double bond endo to the chlorine substituent.
IR spectrum can be used to have a further analysis of electrons distribution by comparing the frequencies of absorbtions. In order to xompare the electron density of endo pi-orbital and exo pi-orbital, the stretching frequency of each double bond should be focused. The stretching frequency of endoalkene is 1757.34 cm-1 while the stretching frequency of exoalkene is 1737.03 cm-1. The higher value the frequency the stronger the bond is, which means the electron density in endo pi-orbital is higher than that of exo pi-orbital. This agrees with the discussion mentioned above.
{kind=link}
{kind=link}
Monosaccharide chemistry and the mechanism of glycosidation
In the Glycosidation reaction( SN2 type mechanism), the stereochemistry of the C-OAc bond controls the stereochemical outcome of the C-Nu bond by nucleophilic attack. This is due to neighbouring-group-participation from the adjacent acetyl group to form a second intermediate oxenium cation. The nucleophile can attack either from the top face to form the β-anomer or the bottom face to form the α-anomer.
Fig8. Mechanisms of potential Glycosidation reaction

There are 4 potential isomers for each of A and B. However, There are a number of O-R groups in the molecule, which can be easily twisted. The conformations of molecules of both A and B presented below are the most stable I could find.
Table8: MM2 and PM6 calculated data for molecule A (Units in kcal/mol)
| Dimer | MM2 Energy | MOPAC/PM6 | Stretch | Bend | Stretch bend | Torsion | Non- 1,4 VDW | 1,4 VDW | Charge/Dipole | Dipole/Dipole |
|---|---|---|---|---|---|---|---|---|---|---|
| Axial | 23.7669 | -86.22435 | 2.5445 | 10.4571 | 0.9776 | 3.1243 | -0.0650 | 19.1116 | -19.1864 | 6.8033 |
| Axial') | 42.5754 | -73.452131 | 2.4305 | 13.3878 | 0.9988 | 2.0715 | -2.7392 | 18.7675 | 1.8229 | 5.8356 |
| Energy Difference(Axial'-Axial) | 18.8085 | 12.772219 | -0.114 | 2.9307 | 0.0212 | -1.0528 | -2.6742 | -0.3441 | 21.0093 | -0.9677 |
| equatorial | 21.1502 | -89.23435 | 2.2317 | 10.6609 | 0.8151 | 1.8296 | -1.3069 | 19.3155 | -16.8289 | 4.4332 |
| equatorial' | 33.0177 | -72.55730 | 2.3429 | 14.1449 | 0.9595 | 0.7459 | -0.7096 | 18.8636 | -11.7037 | 9.3742 |
| Energy Difference(equatorial'-equatorial) | 11.8675 | 16.67705 | 0.1112 | 3.484 | 0.1444 | -1.0837 | 0.5973 | -0.4519 | 5.1252 | 4.941 |
As can be observed from the table and the figure above, the isomer A with axial C-OAc in conformation where the double bond oxygen in the C-OAc is closer to the oxygen ions (Axial) is stable than the other conformation by 18.8 kcal/mol. The main contribution is from the Charge/Dipole, this agrees with the expectation from the difference of the structures. The closer the double bond oxygen to the oxygen ion, the more stable of Charge/Dipole energy. The isomer with equatorial C-OAc is more stable than equatorial', the main contributions are from both Charge/Dipole and Dipole//Dipole. The reason of different of stability is similar as that of "axial" isomers. However, the reason why Dipole/Dipole contributes more in equatorial situation is complicated to analyse here.
The difference of MOPAC energy between equatorial is higher than that of axial, while the difference MM2 between of equatorial is lower than that of axial. This could be explained by the reason that the MOPAC calculation will conclude the factors of electron interaction. Hence, if energy calculations fails in one method, the other method can be replace to have a try.
From the data analysis above, the distances between double bond oxygen and the oxygen ion of axial and equatorial can be determined to be shorter than those of axial' and equatorial'. As can be seen in figure 8, axial and equatorial' tend to form a molecule B with a 'flat' 5-member ring, while axial and equatorial' tend to form a 'flipped' 5-member ring. The energy data of molecule B are shown in below.
Table9: MM2 and PM6 calculated data for molecule B (Units in kcal/mol)
| Dimer | MM2 Energy | MOPAC/PM6 | Stretch | Bend | Stretch bend | Torsion | Non- 1,4 VDW | 1,4 VDW | Charge/Dipole | Dipole/Dipole |
|---|---|---|---|---|---|---|---|---|---|---|
| Axial | 26.6185 | -85.22715 | 1.9268 | 12.9148 | 0.6569 | 7.8805 | -2.6532 | 17.9868 | -10.8857 | -1.2085 |
| Axial' | 40.0723 | -78.55231 | 2.5847 | 16.6185 | 0.7162 | 6.9298 | -2.6954 | 19.3906 | -2.5981 | -0.8740 |
| Energy Difference(Axial'-Axial) | 13.4548 | 6.67484 | 0.6579 | 3.7037 | 0.0593 | -0.9507 | -0.0422 | 1.4038 | 8.2876 | 0.3345 |
| equatorial | 34.5322 | -78.22435 | 1.7481 | 16.0821 | 0.6971 | 7.7595 | -4.0474 | 17.6919 | -4.9953 | -0.4038 |
| equatorial' | 44.0260 | -72.55730 | 2.7135 | 17.4631 | 0.7955 | 8.1687 | -2.4405 | 19.3992 | -0.3281 | -1.7453 |
| Energy Difference(equatorial'-equatorial) | 9.4938 | 5.66705 | 0.9654 | 1.381 | 0.0084 | -0.4092 | 1.6069 | 1.7073 | 4.6672 | -1.3415 |
As can be observed from the table and figure above, the isomer B from axial and equatorial are more stable than axial' and equatorial' by 13.4548 kcal/mol and 9.4938 kcal/mol respectively. The main contributions of both isomers are from Charge/Dipole. MOPAC has the same trend as MM2 in this case.
As can be observed in the figure 8. As the axial (lower energy confomation) and equatorial'(higer enery conformation) molecule A tend to form isomers molecule B with a 'flat' member ring, so nucleophile will attack axial from bottom face to form alpha anomer, while nucleophine will attack equatorial' from up face to form beta anomer. In contrast, axial' (higher energy conformation) and equatorial'(lower energy conformation) molecule A tend to form isomers molecule B with a 'flipped' member ring, so nucleophile will attack axial' from up face to form beta anomer, while nucleophine will attack equatorial from bottom face to form alpha anomer.
Mini-Project: Simulation of spectroscopic data for a literature molecule
Molecule 18 Spectra comparison
The molecules 17 and 18 are derivatives of 9 and 10 shown above, Here the simulation of the 1H and 13C spectra of molecule 18 was conducted, and compared with the literature values.
Fig10, Images of molecule17 and 18

Set-up
1.MM2 Minimisation energy Calculation
2.Input file creation
Job type: Minimise
Method: DFT/B3LYP
Basis set: 6-31G, d,p polarisation
Solvation model: CPCM/ chloroform
3.Gjf file creation
First line of gjf file:# RB3LYP/6-31G(d,p) Opt SCRF=(CPCM,Solvent=chloroform)
4.The calculation was submitted to the HPC, checkpoint file download
First line of gjf file: # mpw1pw91/6-31G(d,p) NMR SCRF=(CPCM,Solvent=chloroform)
Results
The MM2 minimisation energy obtained was 66.0526 kal/mol.
Table11. c13 NMR Data of molecule 19
| Shift (ppm) | Degeneracy | Atoms | Literature results | Difference |
|---|---|---|---|---|
| 218.72 | 1 | 10 | 211.49 | 7.23 |
| 148.78 | 1 | 4 | 148.72 | 0.06 |
| 122.57 | 1 | 6 | 120.9 | 1.67 |
| 89.83 | 1 | 15 | 74.61 | 15.22 |
| 56.67 | 1 | 11 | 60.53 | -3.86 |
| 55.27 | 1 | 3 | 51.3 | 3.97 |
| 54.72 | 1 | 12 | 50.94 | 3.78 |
| 49.75 | 1 | 7 | 45.53 | 4.22 |
| 48.04 | 1 | 16 | 43.28 | 4.76 |
| 45.62 | 1 | 21 | 40.82 | 4.80 |
| 43.65 | 1 | 22 | 38.73 | 4.92 |
| 41.68 | 1 | 18 | 36.78 | 4.90 |
| 38.65 | 1 | 5 | 35.47 | 3.18 |
| 31.24 | 1 | 13 | 30.84 | 0.4 |
| 28.14 | 1 | 2 | 30 | -1.86 |
| 27.86 | 1 | 19 | 25.56 | 2.30 |
| 26.54 | 1 | 8 | 25.35 | 1.19 |
| 25.68 | 1 | 1 | 22.21 | 3.47 |
| 23.19 | 1 | 9 | 21.39 | 1.40 |
| 21.84 | 1 | 17 | 19.83 | 2.01 |
Table12, H NMR Data of molecule 18
| Shift (ppm) | Degeneracy | Atoms | Literature results | Atoms |
|---|---|---|---|---|
| 5.99 | 1 | 31 | 5.21 | 1 |
| 3.66 | 1 | 38 | 3.00-2.70 | 6 |
| 3.55 | 1 | 51 | ||
| 3.30 | 2 | 52,50 | ||
| 3.17 | 1 | 53 | ||
| 3.021 | 1 | 29 | ||
| 2.709 | 1 | 41 | 2.70-2.35 | 4 |
| 2.52 | 4 | 39,24,42,40 | ||
| 2.17 | 1 | 27 | 2.20-1.70 | 6 |
| 2.05 | 2 | 28,26 | ||
| 1.94 | 1 | 25 | ||
| 1.88 | 1 | 45 | ||
| 1.74 | 2 | 44,48 | 1.58 | 1 |
| 1.54 | 5 | 34,37,30,49,43 | 1.50-1.20 | 3 |
| 1.36 | 2 | 47,46 | 1.07 | 3 |
| 1.24 | 1 | 36 | ||
| 1.03 | 3 | 35,32,33 | 1.03 | 3 |
{kind=link}
From the comparison of values of calculations with that from the literature[], The values chemical shifts of both C13 and H are generally close to the values in the literature. C15 which connects with 2 S atoms can be easily pointed out, because it's value is different from that from literature by 15.22 ppm. This can be explained by the concept that carbons attached to "heavy" elements have shifts which need correction for so-called Spin-orbit coupling errors. The general value difference from literature could be explained by the conformation used is not in the lowest energy.However, the difference can be bared because a number of limitations for the comparison. Firstly, the solvent used in literature is different from the solvent in the instruction( C6D6 was used to conduct repeated calculation , the results obtained were similar). Secondly, the exact positions of carbon atoms are not provided in the literature. Lastly, the values of H are provided as a number of ranges.
Analysis of the choice literature molecule chosen
Asymmetric Michael addition of chiral 2-fluoroenaminoesters derived from (S)-1-phenylethylamine to R-substituted methyl acrylate leads to diastereomeric γ-substituted γ-fluoroglutamate precursors.The tertiary center bearing the amino acid function in its natural configuration is generated with a high level of stereocontrol in contrast to the quaternary carbon center. Diastereomeric γ-substituted γ-fluoroglutamates were efficiently separated and isolated as thioketal derivatives harboring very good enantioselectivity.[6]
Fig11. Reaction scheme of molecule5 and 6

The molecule 5 and 6 are isomers, MM2-forcefield was used to determine the minimisation energy of both isomers. The isomers 5 and 6 have relatively large numbers of different conformations due to a number of single bonds can be twisted, 3 pairs of conformations' MM2 minimisation energy are presented here( limited time to conduct all of the conformations), and the conformations of molecule 5 and 6 with the lowest energy was used to do later analysis. The procedures of set-up were followed the lab instructions provided.( The molecule chosen seems to be complicated, however the exact potions of Carbon atoms in C13 NMR are provided, which is useful for comparison.)
| Dimer | MM2 Energy |
|---|---|
| 5 Conformation1 | 24.6170 |
| 6 Conformation1 | 27.8221 |
| 5 Conformation2 | 35.2673 |
| 6 Conformation2 | 40.0671 |
| 5 Conformation3 | 65.7669 |
| 6 Conformation3 | 31.5754 |
| Molecule | MM2 Energy | Stretch | Bend | Stretch bend | Torsion | Non- 1,4 VDW | 1,4 VDW | Dipole/Dipole |
|---|---|---|---|---|---|---|---|---|
| 5 Conformation1 | 24.6170 | 3.0249 | 10.3308 | 0.7561 | -0.5084 | -8.5880 | 13.7808 | 5.8208 |
| 6 Conformation1 | 27.8221 | 2.8914 | 12.7970 | 0.8295 | 0.2758 | -8.5341 | 13.1190 | 6.4434 |
| Energy Difference(M6-M5) | 3.2051 | -0.1335 | 2.4662 | 0.0734 | 0.7674 | 0.0539 | - 0.6618 | 0.6226 |
NMR Analysis
| Shift (ppm) | Degeneracy | Atoms | Literature results |
|---|---|---|---|
| 165.39 | 1 | 8 | 171.68 (COOMe) |
| 164.89 | 1 | 12 | 169.68 (NHCOCH3) |
| 145.60 | 1 | 21 | 5.4657 168.94 (COOEt) |
| 103.21 | 1 | 7 | 100.98 (CqF) |
| 76.93 | 1 | 5 | 70.98 (CH3CS2) |
| 61.76 | 1 | 16 | 62.27 (COOCH2-CH3) |
| 55.06 | 1 | 11 | 52.54 (COOCH3) |
| 52.31 | 1 | 20 | 49.13 (CH) |
| 45.11 | 1 | 2 | 40.20 + 40.72 (SCH2CH2S) |
| 44.07 | 1 | 1 | 35.90 (CH2CH) |
| 43.41 | 1 | 10 | 28.97 (CH3CS2) |
| 33.30 | 1 | 6 | 22.90 (NHCOCH3) |
| 14.75 | 1 | 17 | 13.99 (COOCH2CH3) |
| Shift (ppm) | Degeneracy | Atoms | Literature results |
|---|---|---|---|
| 167.50 | 1 | 8 | 171.68 (NHCOCH3) |
| 162.18 | 1 | 12 | 169.73 (COOMe) |
| 145.48 | 1 | 21 | 169.22 COOEt |
| 99.67 | 1 | 7 | 100.01 (CqF) |
| 75.71 | 1 | 5 | 71.05 CH3CS2 |
| 61.71 | 1 | 16 | 62.11 (COOCH2-CH3) |
| 54.77 | 1 | 11 | 52.38 (COOCH3) |
| 51.74 | 1 | 20 | 48.18 (CH) |
| 44.89 | 1 | 2 | 40.11+40.64 (SCH2CH2S) |
| 41.39 | 1 | 1 | 36.18 (CH2CH) |
| 40.84 | 1 | 10 | 28.67 (CH3-CS2) |
| 28.98 | 1 | 6 | 22.75 (NHCOCH3) |
| 14.42 | 1 | 17 | 13.83 (COOCH2CH3) |
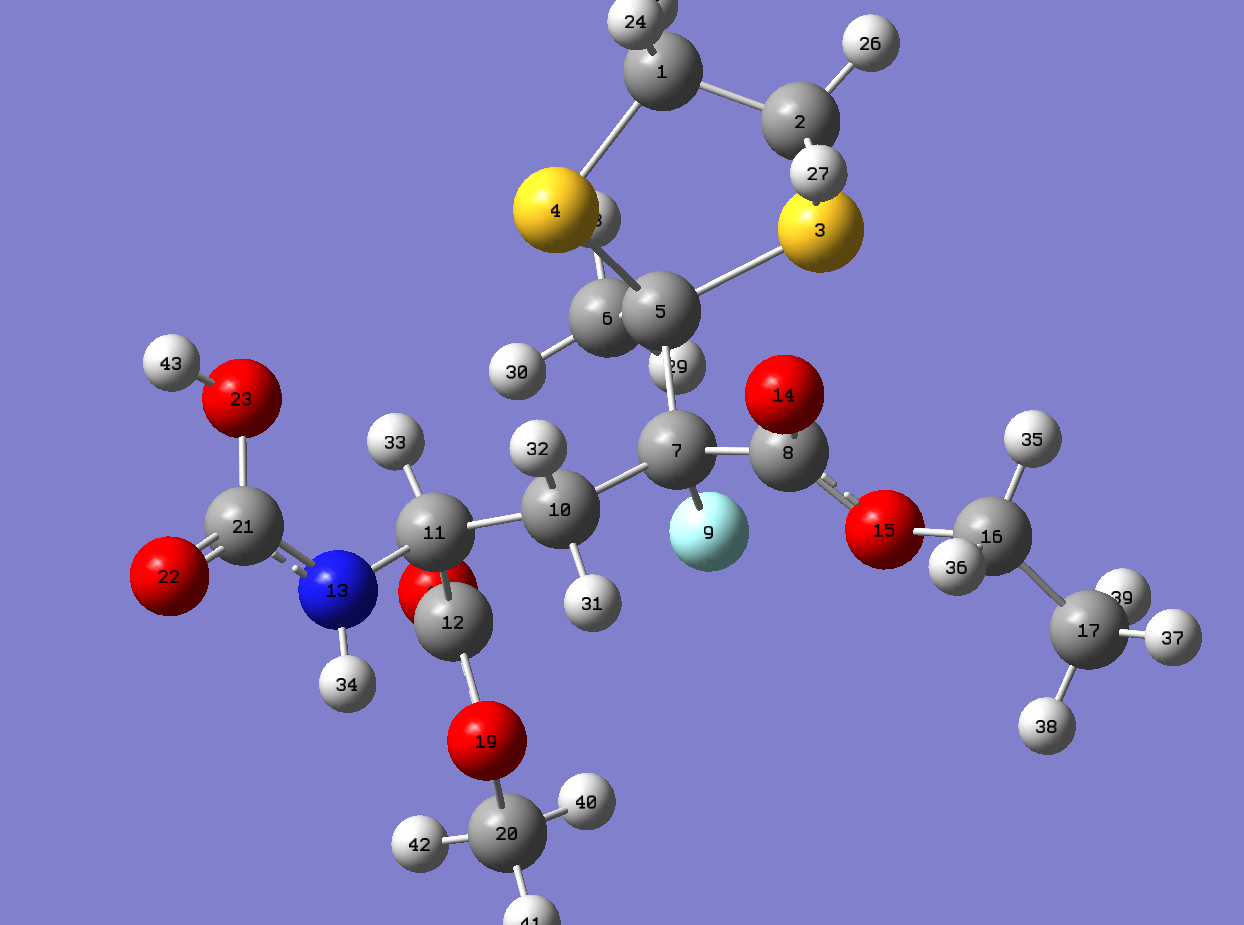{kind=link}
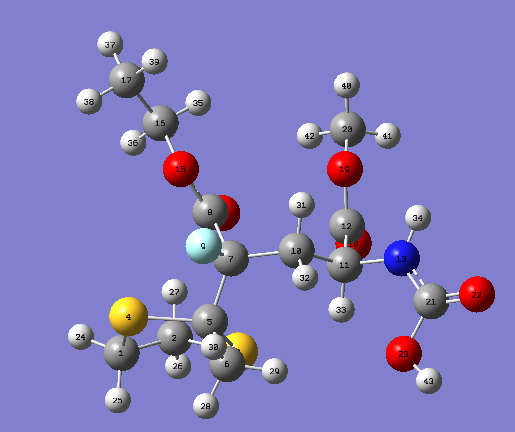{kind=link}
From the comparison of values of calculations with that from the literature[6], The values chemical shifts of C13 of both molecule 5 and 6 are generally close to the values in the literature.However, There are a number of obvious difference can be pointed put.Firstly, the values of C connected with 2 S atoms can be easily pointed out, this can be explained by the concept that carbons attached to "heavy" elements have shifts which need correction for so-called Spin-orbit coupling errors. Secondly, the highest chemical shifts of both molecule 5 and 6 are the carbon in(COOMe). In contrast, in literature,the highest chemical shifts are the carbon in (COOMe) in molecule 5 and the carbon connected with double bond Oxygen(NHCOCH3)in molecule 6. Lastly, a number of low chemical shifts have different orders compared with literature in both molecule 5 and 6. The last two points of difference can be explained by the conformations used may not in the lowest energy.The Number of conformations of this molecule is large, different conformations of branched groups could lead to various chemical shift. For example, the branched carbon get closer to the electronegative atom such as Oxygen in other groups will have large chemical shifts. Moreover, in some case, the chemical shifts are contributed from a combination of conformations of a molecule instead of the one with lowest energy.
IR Analysis
{kind=link}
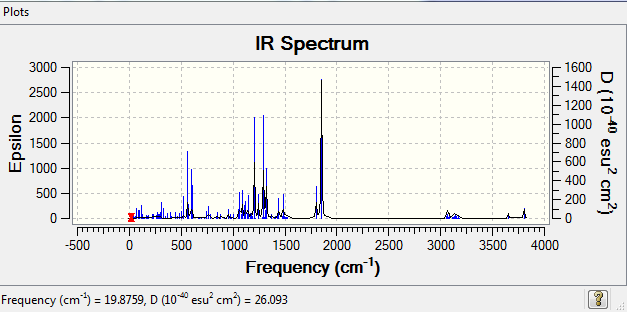{kind=link}
As can be observed from the images above, the IR spectra of molecule5 and 6 have similar distributions with different intensity. The distribution of IR spectrum mainly depends on the functional groups in the molecule, molecule 5 and 6 are isomers, so the spectra are expected to have similar distributions, The intensity depends on the selection rules, which means it depends on the symmetry of molecules. Hence of the intensities are different. There pairs of vibrations are chosen below.
| Molecule5 No. of modes | Frequency/cm-1 | Intensity | Animation | Molecule6 No. of modes | Frequency/cm-1 | Intensity | Animation |
|---|---|---|---|---|---|---|---|
| 38 | 548.85 | 16.99 | Molecule5 | 38 | 561.27 | 99.72 | Molecule6 |
| 78 | 1297.16 | 170.58 | Molecule5 | 78 | 1290.4 | 251.74 | Molecule6 |
| 81 | 1329.06 | 12.94 | Molecule5 | 81 | 1319.89 | 174.61 | Molecule6 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The different of frequencies could be explained by the the steric effect by the surrounding groups. The harder the bonds vibrates, the higher the frequency will be. Molecule6 has generally higher intensity than that of molecule5, this could be determine molecule 5 has relatively symmetric structure.
Optical Rotation (OR) Analysis
| Isomer | Optical rotation/degrees |
|---|---|
| Molecule 5 | 14.28 |
| Molecule 6 | -54.95 |
The magnitude of optical rotation of molecule5 and 6 are different. This agrees with the expectation, the magnitudes of isomers will be the same when they are entanisomers(mirror images to each other). They are two chiral centers in these molecules, they will be entantisomers only when both of two chiral centers are changed to the opposite ones. From the calculation, these isomers can be determined as stereoisomers.
Electronic Circular Dichroism (CD/UV-Visible) Spectrum Analysis
CD AND UV spectra of molecule5
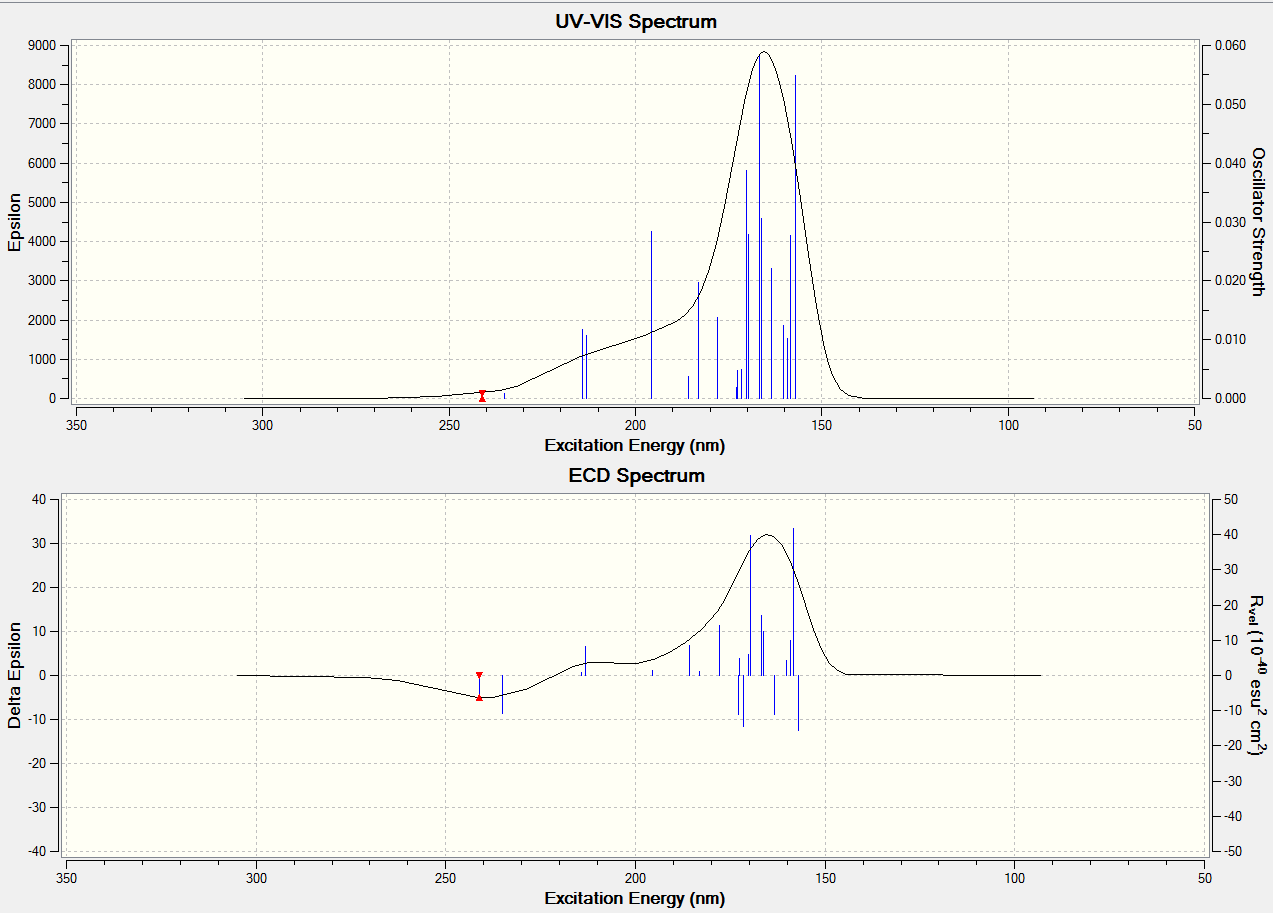{kind=link}
CD AND UV spectra of molecule6
{kind=link}
Both of the related CD AND UV Spectra of molecule 5 and molecule 6 are similar, this could be used to determine this pair of isomers have same colour shown.
Conclusion
From the C13 NMR analysis, the values of calculation is generally consistent with those of literature. This means the conformations of isomers used for calculations are almost in the lowest energy. IR and OR have also successfully confirm the existence of isomers.
References
1.DOI: 10.1021/ja00947a033
2.D. M. Whitfield, T. Nukada, Carbohydr. Res., 2007, 342, 1291.
3.DOI: 10.1021/ja00004a040
4. B. Halton, R. Boese and H. S. Rzepa., J. Chem. Soc., Perkin Trans 2, 1992, 447.
5. DOI: 10.1021/ja00157a043
6. DOI: 10.1021/jo101417c