Module 1: Structure and Spectroscopy (Molecular Mechanics and Molecular Orbitals)
For the purposes of this module ChemBio3D Ultra and Gauss View were used in conjunction with the Imperial College SCAN server to model a series of molecules, predict molecular orbitals and predict spectra.
Part 1: Exercises
The Hydrogenation of Cyclopentadiene Dimer
The cyclopentadiene dimers described in table 1 were defined using the modeling software ChemBio3D Ultra. The MM2 force field that is preset on this software was then used to optimise the molecules geometry.
Table 1:Cyclopentadiene Isomers
|
| 1
|
2
|
3
|
4
|
Cyclopentadiene Endo Dimer | | |
|
Cyclopentadiene Endo Dimer | | |
|
Cyclopentadiene Endo Dimer | | |
|
Cyclopentadiene Endo Dimer | | |
|
| Minimum Energy=35.0271 kcal/mol
|
Minimum Energy= 31.8834 kcal/mol
|
Minimum Energy=35.9319 kcal/mol
|
Minimum Energy= 31.1624 kcal/mol
|
The MM2 force field is a molecular mechanics model first described by Allinger in 1977, the program works by calculating the energy of a molecule in terms of a set of classical potential energy functions. In this approach atoms that are more than two atoms apart have interactions governed by van der Waals, steric repulsion and electrostatics whereas atoms that are directly bonded have interactions described by [Hooke's Law] since bonds are modelled as springs. ChemBio3D Ultra uses a modified version of Allinger's original program that takes account of some additional factors to improve the original force field. The force field used gives a total potential energy based on the following summation:
ETOT=EBEND+ETORSION+ESTRETCH+ENON-BONDED
The Hydrogenation of Cyclopentadiene Dimer Continued
The dimerisation is an example of a (4+2)-cycloaddition reaction more commonly referred to as a Diels-Alder reaction. In which one molecule of cyclopenadiene acts as a 4p electron diene and the second a 2p electron dieneophile. The endo (1) and exo (2) cyclopentadiene dimers were minimised using the MM2 and their relative energies can be seen in table 1. From these results it can be seen that the exo (2) dimer is thermodynamically the most stable as it is closest to a 'staggered' conformation. Since this dimer is not predominant in reactions these results suggest that the reaction proceeds under kinetic control. This is due to a concerted cyclic transition state in which orbitals align more favourably for the endo isomer that the exo isomer.
The hydrogenation of the endo dimer leads to two possible products, since either of the double bonds in the molecule can be hydrogenated. This leads to two different molecules 3 and 4. These molecules were also defined on ChemBio3D Ultra and the energies minimised and calculated using the MM2 force field option. The relative energies of these molecules can also be seen in table 1. From the total energies it is clear that molecule 4 is the most thermodynamically stable and therefore the most likely product of hydrogenation in terms of thermodynamics. Comparissons of all the components that sum to give the total potential energy under the MM2 force field option show significant differences in the bending energy, the difference in the bending energies was 4.3414 kcal/mol which accounts for almost all of the total difference in total potential energy between the two molecules.
Stereochemistry of Nucleophilic Additions to a Pyridinum Ring (NAD+ Analogue) [3]
The reactant for the reaction shown below was defined using ChemBio3D Ultra, the Grignard reagent used in the reaction could not be included in the model as the program did not recognise the magnesium metal. The nucleophile could be included by adding a CH3-ion which is effectively the nucleophile in the reaction, no calculations could however allow for the coordination of the magnesium and therefore the nucleophile will be excluded from the model. The total energy of the reactant was found by MM2 force field minimisation to be 25.5168 kcal/mol. The angle between the oxygen and the ring was found after initial minimisation to be 22.4o, this means the oxygen is pointing above the plane of the aromatic ring system. The oxygen angle was then altered and the molecule was again minimised, in this instance the angle was found to be 17.0o and the energy was considerably higher at some 167.7928 kcal/mol. A more detailed evaluation of the energies used in the MM2 force field showed that the most significant contribution to this energy came from the bending energy (116.549 kcal/mol), this is also signigicantly higher than the bending energy of the 22.4o example. A dramatic potential energy is also observed when the bond angle is altered to -162o, in this instance the energy is found to be 167.6235 kcal/mol. Again the bending energy accounts primarily for this large difference in energy in comparisson to the 22.4o case. These changes in bending energy are most likely due to the way the molecule was manipulated on the screen to give the different dihedral angles. More importantly for the purposes of this project is the change in 1,4-van der Waals interaction energy in the potential energy function. The van der Waals interaction energy doubles as the bond angle was changed from 22.4o to 17.9o or as the oxygen eclipses the hydrogen in the aromatic system. This suggests that the oxygen is positioned in such a way as to be angled up relative to the ring in order to prevent eclipsing and hence give a lower energy conformer. In all cases the oxygen and nitrogen are in a planar arrangement this maybe due to delocalisation in the amide.
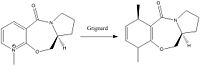
Nucleophilic Addition to Pyridinum Ring Reaction[4]
|
|
The reaction shown above proceeds to give 6 with absolutely all molecules having the stereochemistry depicted. This is due to the position of the oxygen in the reactant, which optimally minimises in such a way that it is angled up relative to the aromatic system, when the magnesium coordinates to the oxygen the methyl anion is effectively forced to attack from above the plane of the pyridine ring. This leads to only one possible stereochemistry for the product in which the methyl group is positioned as shown.
Molecule 6
|
Cyclopentadiene Endo Dimer | | |
|
| 1,4 van der Waals=22.3439 kcal/mol
|
| Bend = 122.3821 kcal/mol
|
| Total Energy= 184.3666 kcal/mol
|
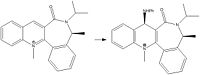
The reaction shown below shows the nucleophilic attack of a second pyridinum ring system (7) by phenyl amine (8). For the purposes of these intial calculation the phenyl amine has been excluded. After the initial minimisation 7 had a dihedral angle of -42.6o the energy was found to be 16.6210 kcal/mol in this case the oxygen is pointing below the plane of the ring the angle was then manipulated and the results can be seen in table 2. From table 2 it can be seen that the most stable confomrers exist when the oxygen is positioned so that it is pointing down relative to the aromatic system. Again the lower energy conformations are those that have the smallest 1,4-van der Waals interaction. This suggests that there is a 1,4-van der Waals interaction between the oxygen and neighbouring hydrogen of the aromatic system. Again the carbonyl is coplanar with the nitrogeon of the amide group.
Reaction of Second Pyridinum Ring System [4]
|
|
Table 2: Modelling of Molecule 7
|
Cyclopentadiene Endo Dimer | | |
|
Cyclopentadiene Endo Dimer | | |
|
| 1,4 van der Waals= 17.562 kcal/mol
|
1,4 van der Waals= 17.617 kcal/mol
|
| Total Energy= 15.4680 kcal/mol
|
Total Energy= 15.1570 kcal/mol
|
The stereochemistry of the product is most likely due to lone pair repulsion. There is a lone pair on the carbonyl oxygen and a lone pair on the aniline, these lone pairs will repel each other as they move closer together in space. This repulsion will force the aniline to attack from the top face of the aromatic system as this mode of attack will reduce the lone pair-lone pair repulsion and hence the stereochemistry of 8 is as shown in the reaction scheme.
Stereochemistry and Reactivity of an Intermediate in the Synthesis of Taxol
Molecules 10 and 11 are different conformations of a key intermediate in the synthesis of taxol an antineoplastic drug used in the treatment of ovarian cancers [5]. The structures of the intermediate were first defined using ChemBio 3D Ultra and then minimised using the MM2 force field the conformers and their relative energies can be seen below. From the results it is clear that the atropisomer with the oxygen pointing up is the least stable suffering from greater torsional strain.
Conformers of a Taxol Intermediate[6]
|
| 10
|
11
|
Cyclopentadiene Endo Dimer | | |
|
Cyclopentadiene Endo Dimer | | |
|
| Minimum Energy= 45.3642 kcal/mol
|
Minimum Energy= 54.9451 kcal/mol
|
The alkene in 10 reacts abnormally slowly to prevent the addition of a further 2 sp3 carbon centres being added to the ring and leading to further strain. This is demonstrated the energy of 10 with a hydrogenated alkene which after MM2 minimisation was found to be 59.781 kcal/mol in the hydrogenated case and 45.387 kcal/mol in the same conformation taxol. It is also worth noting that the alkene is very hindered to electrophilic attack due to the bridging group.
How One Might Induce the Room Temperature Hydrolysis of a Peptide
Molecules 13 and 14 were defined using ChemBio 3D Ultra after MM2 minimisation of the structures the energies were recorded. These energies can be seen in the table below, many different starting points were trialed for the minmisation in order to achieve as low an energy as possible. The position (axial or equatorial)of the nitrogen substituent with respect to the decalin ring was also altered. In all cases a chair-chair conformation was used for the decalin ring in order to ensure the lowest possible energy conformation.
Models of Molecule 13
|
| Di-Equatorial
|
Equatorial O Axial Ethylamido
|
Di-Axial
|
Axial O Equatorial Ethylamido
|
Cyclopentadiene Endo Dimer | | |
|
Cyclopentadiene Endo Dimer | | |
|
Cyclopentadiene Endo Dimer | | |
|
Cyclopentadiene Endo Dimer | | |
|
| Total Energy= 23.2763 kcal/mol
|
Total Energy= 26.2673 kcal/mol
|
Total Energy= 30.3799 kcal/mol
|
Total Energy= 26.3011 kcal/mol
|
Models of Molecule 14
|
| Equatorial Ethylamido Axial O
|
Di-Axial
|
Cyclopentadiene Endo Dimer | | |
|
Cyclopentadiene Endo Dimer | | |
|
| Total Energy= 15.4166 kcal/mol
|
Total Energy= 23.5035kcal/mol
|
It is clear from the results that the di-equatorial conformer of molecule 13 is the more stable as it has the lowest energy following MM2 minimisation. The literature [7] quoted a difference of 5.0 kcal/mol between the diaxial and diequatorial conformers. Comparisson with differences obtained above shows a percentage difference of 71% which shows low correlation between the literature and achieved value. Both reactions are of order one and intramolecular therefore the reactions proceed faster than under normal conditions. Molecule 13 reacts more rapidly than molecule 14 this is because the equilibrium betweeen conformations for molecule 13 lies to side of the di-equatorial conformer, which is the most reactive conformer. In molecule 14 however the equilibrium lies to the side of the equatorial ethylamido and axial oxygen which is the least reactive. This means that an energy barrier has to be overcome for the hydrolysis of 14 to proceed. Hydrogen bonding is a factor in these reactions as it helps to hold the reacting groups in there most reactive position.
Modelling Using Semi-Empirical Molecular Orbital Theory
This method is more advanced than the previously employed molecular mechanics method as it takes account of molecular orbital theory and the interaction of all of the electrons in the molecule.
Regioselective Addition of Dichlorocarbene
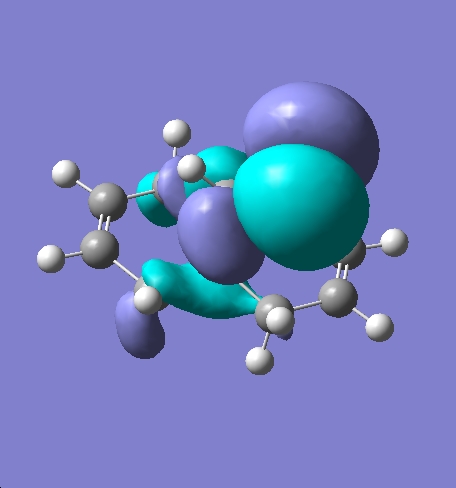
Molecule 15 was first defined and minimised using ChemBio 3D Ultra and MM2 the file was then exported to GaussView in the .gjf format. Once opened in GaussView the molecule was optimised again using the Gauss View calculation appelet with a DFT method employing the B3LYP/6-31G basis set. The check point file was then opened in the MO editor pane and various MOs visualised. These MOs can be seen below, the HOMO, HOMO-1, LUMO, LUMO+1 and LUMO+2 are displayed.
MOs of Molecule 15
|

|
|

|

|

|
| HOMO
|
HOMO-1
|
LUMO
|
LUMO+1
|
LUMO+2
|
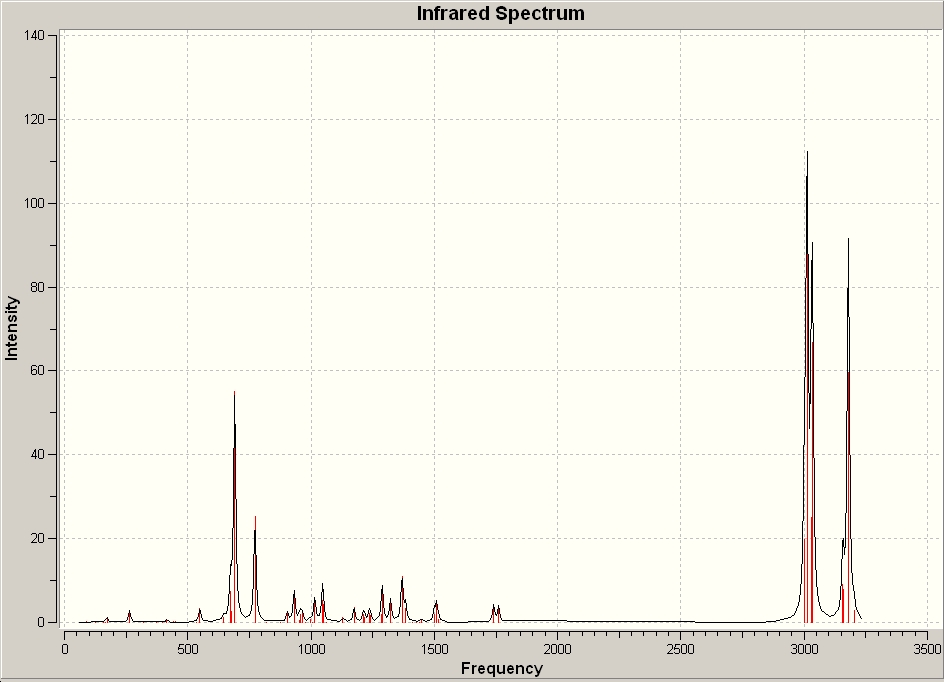
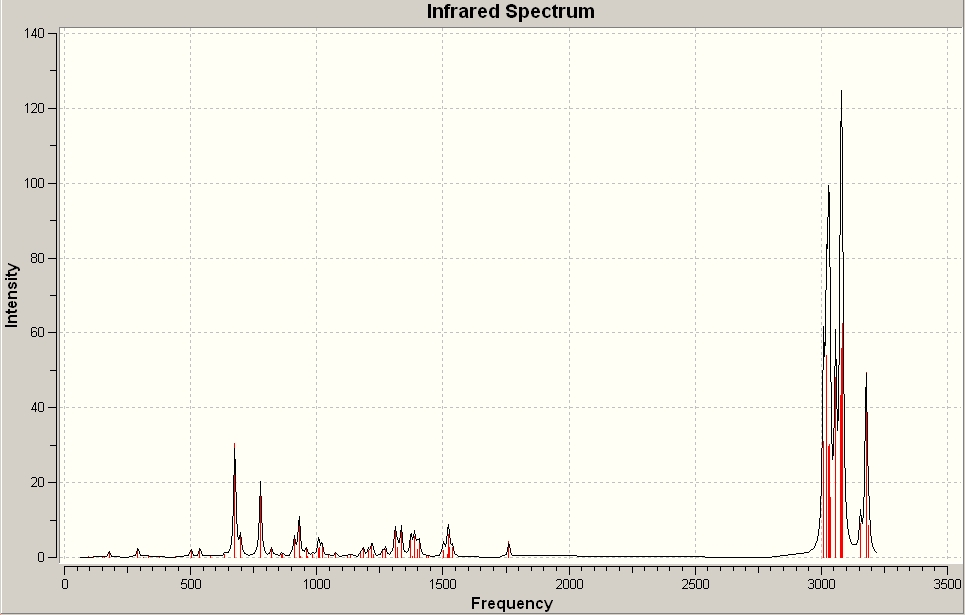
An electrophile such as dichlorocarbene will attack 15 at the double bond endo to the chlorine in the bridging group. This is because the pi-orbital of the exo double bond can interact with the sigma anti-bonding orbital of the C-Cl bond. This serves to reduce electron density at the exo double bond and increase electron density on the C-Cl bond, this in turn leads to a weaker more delocalised C-Cl bond and a less nucleophilic endo double bond hence electrophilic attack will occur at the endo double bond [8] A monohydrogenated molecule 15, molecule 16, was then defined using ChemBio 3D Ultra this molecule was then minimised using MM2 before being exported to Guassian and submitted to SCAN for further optimisation. Once the optimisation was complete both molecule 15 and 16 were submitted to SCAN running an optimisation and frequency calculation to return an infra-red spectrum. The spectra can be seen below:
Molecules 15 and 16 Optimisation and Spectra
|
Cyclopentadiene Endo Dimer | | |
|
Cyclopentadiene Endo Dimer | | |
|
|
|
Since the wavenumber of a peak in an infrared spectrum is directly proportional to the square root of force constant and force constant is effectively bond strength the arguements made above can be rationalised in terms of IR. In molecule 15 there is an interaction between the endo double bond pi orbital and the C-Cl sigma bond antibonding orbital. This has two effects 1) the C-Cl bond is weakened by the increased electron density and 2) the C=C bond is weakend by the removal of the electron density. This means that in the IR spectra the C-Cl stretch for molecule 15 should appear at a lower wavenumber than the C-Cl stretch for molecule 16, furthermore the endo C=C stretch should appear at a lower intensity than the C=C exo stretch in moleculoe 15. Both of these predictions were confirmed by the predicted IR spectra the C=C exo stretch in molecule 15 appears at 1760.97cm-1 and the endo stretch at 1740.78cm-1. In molecule 15 the C-Cl stretching frequency occurs at 772.604cm-1 and in molecule 16 the same stretch occurs at 776.81cm-1.
Part 2: Mini-Projects
Assigning Regioisomers in "Click Chemistry"
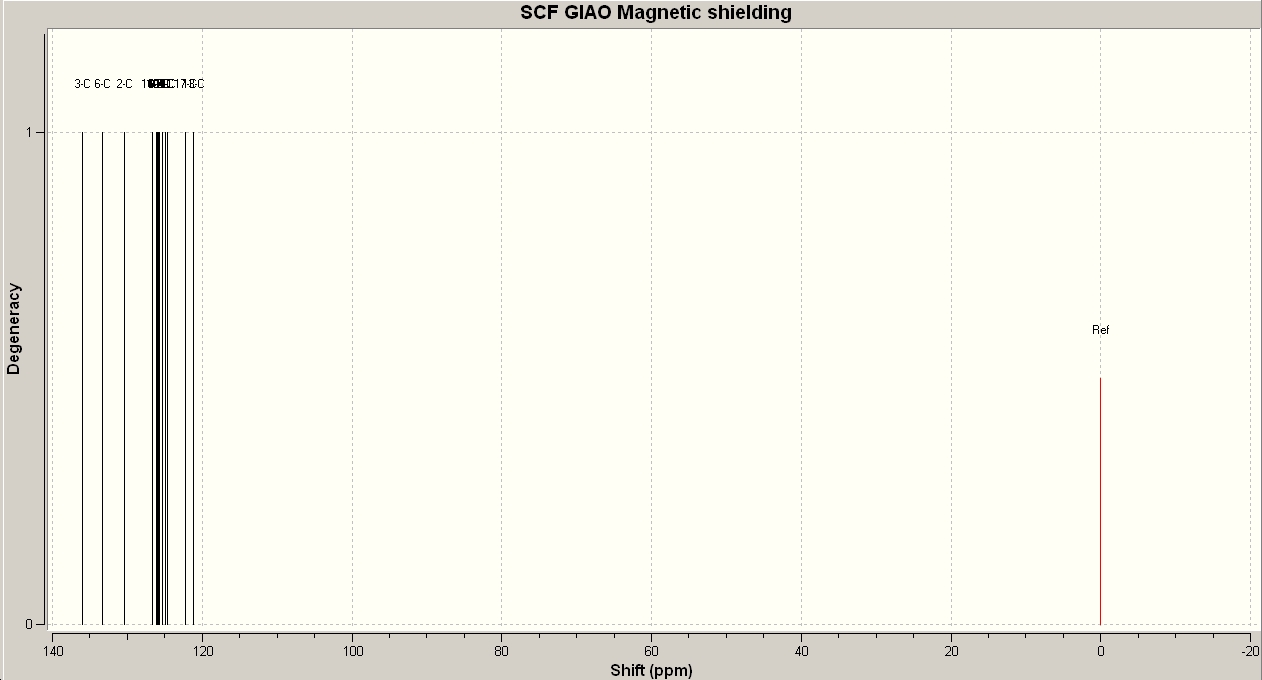
The reaction given below proceeds to form either product A or B depending on the catalyst used, isomer A (a 1,4-somer) is formed using a Ruthenium catalyst whereas isomer B (a 1,5isomer) is formed via a Copper catalyst [9] . Both isomers A and B were defined using ChemBio 3D Ultra and were then minimised using MM2. After the MM2 minimisation the molecules were exported into Gaussian and minimised using the 6-31G basis set. The Gaussian input files were then submitted to SCAN for DFT NMR calculations. A CDCl3 reference solvent was added. 13CNMR is the principle spectroscopic method sued to resolve the different possible products and the NMR produced by calculation can be seen below.
Click Chemistry NMR
|
| A
|
B
|
Cyclopentadiene Endo Dimer | | |
|
Cyclopentadiene Endo Dimer | | |
|

|
|
|
|
These 13C-NMR predicted spectra are in close correlation to the reported 13C-NMR data published in the literature[9]. Table (i) summarises the key differences and similarities in the the NMRs predicted and reported. The key result is for the carbon labelled 3 in molecule B and 2 in molecule B (marked in red on diagrams in table (i)) give the key to resolving the structure. In the A isomer the nitrogen and phenyl aromatic systems are in the same plane and therefore have a large effect on the chemical shift of the carbon attached to the phenyl system, in the B isomer however the aromatic systems are not planar and therefore the aromaticity and conjugation is reduced, and hence the chemical shift is lower. An IR spectrum of both products was also produced, but there was no distinguishable differences between the molecules. This is because the bonding in bot A and B is similar.
Table ii:13C NMR Literature [10] Comparisson for A
|
| Literature δ/ ppm
|
119.5
|
125.5
|
127.9
|
128.56
|
128.64
|
128.9
|
130.4
|
134.6
|
148.0
|
| Model δ/ ppm
|
115.037, 115.637
|
121.632, 122.231
|
124.779
|
125.379, 125.275, 125.244
|
126.278
|
126.728
|
127.177
|
133.922
|
144.713
|
Table ii:13C NMR Literature [9] Comparisson for B
|
| Literature δ/ ppm
|
126.93
|
127.22
|
128.22
|
128.92
|
129.08
|
129.64
|
133.26
|
133.34
|
135.66
|
138.26
|
| Model δ/ ppm
|
121.179
|
122.229
|
124.675, 124.856
|
125.384
|
125.759, 125.923
|
126.087
|
126.644, 126.596
|
130.384
|
133.322
|
135.993
|
The data generated by the computational modelling is generally in good agreement with the data from experiment. The modelled data is however of greater resolution and therefore there are more peaks displayed in the modelled data than the experimental data.
Largazole [11]
Largazole is a potent cytotoxic cyclodepsipeptide derived from cyanobacterium that were collected in the florida keys. Largazole has 4 stereocentres giving 16 (24) different possible conformations. For the purposes of this study the difference in energy between the (+) and (-) form of Largazole will be investigated. Both compounds were first defined using ChemBio 3D Ultra and then exported as Gaussian Input Files (.gjf), it was decided that Largazole due to its size and complexity should be optimised by submission to the scan server. The gjf files generated were uploaded to the scan server to be optimised using a 6-31G basis set. The minimised outputs can be seen below.
Largazole Minimised Outputs
|
| (+)-Largazole
|
(-)-Largazole
|
Cyclopentadiene Endo Dimer | | |
|
Cyclopentadiene Endo Dimer | | |
|
| E= -2919.794
|
E= -2920.174 a.u
|
Comparisson of the energies following optimisation shows that largazole + to be the most stable conformation, there is however only a small energy difference. 13C-NMR spectra were also produced and these can be seen below. The optical rotation of the largazole compounds is currently being calculated and this along with a more detailed analysis will be posted as soon as possible
References
- ↑ ChemBio 3D Ultra, Help Information Menu, accessed 25/10/2008
- ↑ Allinger's Molecular Mechanics Research Lab, 1995, http://europa.chem.uga.edu/, Date Accessed: 25/10/2008
- ↑ A. G. Shultz, L. Flood and J. P. Springer, J. Org. Chemistry, 1986, 51, 838 DOI:10.1021/jo00356a016
- ↑ 4.0 4.1 Computational Chemistry Organic Module Lab Script, Imperial College London, http://www.ch.ic.ac.uk/wiki/index.php/Mod:organic, last accessed 31/10/2008
- ↑ BNF56, 2008, www.bnf.org, Date Accessed: 15/10/2008
- ↑ S. W. Elmore and L. Paquette, Tetrahedron Letters, 1991, 319 DOI:10.1016/S0040-4039(00)92617-0 10.1016/S0040-4039(00)92617-0 10.1016/S0040-4039(00)92617-0 10.1016/S0040-4039(00)92617-0 10.1016/S0040-4039(00)92617-0
- ↑ M. Fernandes, F. Fache, M. Rosen, P.-L. Nguyen, and D. E. Hansen, 'Rapid Cleavage of Unactivated, Unstrained Amide Bonds at Neutral pH', J. Org. Chem., 2008, 73, 6413–6416 DOI:10.1021/jo800706y
- ↑ B. Halton, R. Boese and H. S. Rzepa., J. Chem. Soc., Perkin Trans 2, 1992, 447 DOI:10.1039/P29920000447
- ↑ 9.0 9.1 9.2 Zhang and Chen,Ruthenium-Catalyzed Cycloaddition of Alkynes and Organic Azides, J. Am. Chem. Soc. 2005, 127, 15998 DOI:10.1021/ja054114s
- ↑ Sirion and Bae, Ionic Polymer Supported Copper(I): A Reusable Catalyst for Huisgen's 1,3-Dipolar Cycloaddition, Thieme, Supporting Information Used DOI:10.1055/s-2008-1078245
- ↑ Structure and Activity of Largazole, a Potent Antiproliferative Agent from the Floridian Marine Cyanobacterium Symploca sp, Taori and Paul, J. Am. Chem. Soc., 130 (6), 1806 -1807, 2008 DOI:10.1021/ja7110064













