
Rep:Mod:jfg06
Modelling using Molecular Mechanics
Cyclopentadiene
Dimerisation Products
Cyclopentadiene is able to act as both a diene and a dieneophile and therefore readily dimerises by a [2+2 cycloaddition] to give two products; an endo and exo dimer.
-
 Exo Dimer1
Exo Dimer1 -
 Endo Dimer 2
Endo Dimer 2 -
 Exo Dimer 1 (no hydrogens)
Exo Dimer 1 (no hydrogens) -
 Endo Dimer 2 (no hydrogens)
Endo Dimer 2 (no hydrogens)




By minimising the energy of the two dimerisation products at the MM2 level, the endo dimer 2 is found to be 2.12kcal/mol or 0.0034 H higher in energy than the thermodynamically preferred exo isomer 1. The results of the minimisation can be broken down into energy terms dealing with specific molecular deformations. They are shown in the table below.

| Molecule | Exo dimer 1 | Endo dimer 2 | Difference |
|---|---|---|---|
| Stretch | 1.27 | 1.26 | -0.01 |
| Bend | 20.58 | 20.84 | +0.26 |
| Stretch-Bend | -0.84 | -0.84 | 0.00 |
| Torsion | 7.67 | 9.51 | +1.84 |
| Non 1,4-VdW | -1.42 | -1.56 | -0.14 |
| 1,4-VdW | 4.24 | 4.33 | +0.09 |
| Dipole-Dipole | 0.38 | 0.45 | +0.07 |
| Total | 31.88 | 34.00 | +2.12 |
It is clear from the energy minimisation results that the torsion and stretch-bend contributions account for the majority of the energy difference between the two isomers. The torsion term can be explained by considering the two dihedral or "torsion" angles C3-C4-C7-C10 and C1-C2-C6-C8. In the exo isomer 1 these are much greater than in the endo isomer 2, indeed C3 and C1 are trans to C10 and C8 in the former but are cis in the later. This cis-trans difference means that in the endo isomer there is a profound steric clash between the protons on C3 with those on C1 and the protons on C10 with those on C8. Obviously this interaction greatly increases torsional strain and so the torsional energy term in the endo isomer. The bend energy term which "represents the energy associated with deforming bond angles from their optimal values", is larger in the endo isomer for the same reason. Thus the exo isomer is the more thermodynamically stable.
The selective "non-thermodynamic" preference for the endo isomer can be justified if the molecular orbitals of the two cyclopentadiene fragments are considered. Shown by the thumb on the right is a graphic of the MO interactions between the LUMO of the cyclopentadiene behaving as the dieneophile and the HOMO of the cyclopentadiene behaving as the diene. The red dashed lines indicate the bonding interactions which will form the 2 new σ bonds. The black lines show favourable, non-bonding orbital interactions. It is the later which help to stabilise the transition state leading to the endo dimer and therefore skew the ratio of products in favour of the less thermodynamically stable endo product 2.
Further explanation of the so called "endo rule" are-
-symmetric alignment of HOMO LUMO is due to an initial π-π interaction between the two rings followed by rotation around a central axis.
-quantitative research on the MO's involved suggests that large orbital coefficients favour this orientation and so the endo product.
Hydrogenation Products
It is known that initial hydrogenation of the endo product proceeds at either of the two double bonds but not at both alkene moieties. Reduction at one of these sites results in the following products.
-
 Hydrogenation Product 3
Hydrogenation Product 3 -
 Hydrogenation Product 4
Hydrogenation Product 4


After using the MM2 modelling method to minimise their energies, the reduction products 3 and 4 can be compared in the same way as the cyclopentadiene dimers. The results of the energy minimisation (shown below) show that 4 is the more stable product by 4.77 kcal/mol or 0.0076 H.


| Molecule | Hydration Product 3 | Hydration Product 4 | Difference |
|---|---|---|---|
| Stretch | 1.23 | 1.10 | 0.13 |
| Bend | 18.97 | 14.55 | 4.42 |
| Stretch-Bend | -0.76 | -0.06 | -0.70 |
| Torsion | 12.15 | 12.49 | -0.34 |
| Non 1,4-VdW | -1.56 | -1.09 | -0.47 |
| 1,4-VdW | 5.73 | 4.52 | 1.21 |
| Dipole-Dipole | 0.16 | 0.14 | 0.02 |
| Total | 35.93 | 31.16 | 4.77 |
Qualitatively examining the structures of these product we may expect 3 to be of higher energy since it contains a double bond in a bridged ring system and this feature is particularly strained due to compressed alkene bond angles. By examining the quantitative evidence we find that the torsion term accounts for the majority of the energy difference between the two products. Indeed the torsion energy in product 3 is 4.42 kcal/mol or 0.0070 H higher than in product 4. Now considering the alkene moiety, which has an optimum bond angle of 120o, the measured alkene dihedral angles are 108o in product 3 compared to a much more favourable 112.5o in alkene 4. It can therefore be concluded that the majority of the torsional energy difference, and by that argument the overall energy difference, is indeed due to compression of the alkene bond angles from their optimum positions and that product 4 should be the thermodynamically more stable, due to a less strained unsaturated moiety.
The kinetic stability of products depends on the rate of reaction and is therefore governed by the ease of attack of the reductant at the alkene. The double bond in the bridged ring is slightly more obscured (by the bridge) than the other alkene and so we would perhaps expect a slight preference towards product 3 on kinetic grounds.
Stereochemistry of Nucleophilic additions to a pyridinium ring (NAD+ analogue)
Methylation of Prolinol
Reacting the prolinol derivative 5 with a Grignard reagent results in methylation at the 4 position. Reaction occurs here as a prominent resonance form of the reactant places a positive charge at carbon 4.

In order to predict the stereochemistry of the product i.e. if the methyl group is added on the top or bottom face of the ring as the molecule is drawn here, conformations of the molecule must be probed.
The energy of the reactant was minimised with the amide oxygen either equatorial with respect to the aromatic ring or axial. This can be achieved by manually setting the position of the oxygen or positioning the oxygen in such a place that iteration of the minimum energy achieves a local minimum. In the case of the axial conformer the later approach was used and in the case of the equatorial a combination of the two was most appropriate.
N.B. If the calculation is run with the Grignard reagent present the programme throws up an error stating that the atom type is not defined for the Mg atom. The atom type should be 122 from the atomic number 12x10 + 2 ligands =122. This does not appear in the atom type table that ChemDraw 3D refers to but apparently can be added manually if especially required.
The gallery below shows images of the conformers described above. Care was taken to preserve the stereochemistry at the chiral carbon with the hydrogen pointing down since ignoring this can lead to several additional conformers which are comparable in minimum energy to the ones shown here. Where appropriate the dihedral angles are also shown

|

|

|

|
| 5a Axial Up 149.69kcal/mol or 0.23854H | 5b Axial Down 144.18kcal/mol or 0.22977H | 5c Equatorial 25.88kcal/mol or 0.04125H | 5d Equatorial Planar 32.80kcal/mol or 0.05226H |
It is obvious that although the axial conformers are interesting in structure, they are far too high in energy to occur in the reaction, therefore they will not be discussed further. The equatorial conformers however are found as predicted in the literature. Several local minima ranging from 25.8836 kcal/mol to over 32.7953 kcal/mol have been found for the conformer with C=O pointing above the ring. However conformer 5d was artificially created by taking the conformer 5c as a starting point and setting the dihedral angle between the amide oxygen and the ring to 0o. This was done solely on the suggestion of the literature which implied it to be an important energy minimum, this claim can now be disputed since it is over 6kcal/mol higher in energy than the lowest other upward pointing conformer.

It is supposed that during this reaction as the methyl is delivered to the 4 position on the aromatic ring, the carbonyl plays an important role; bonding to the electropositive Mg atom thus forming a stabilised 6 membered transition state. (see above) If the geometry about the Mg is linear, or bent, and the iodine is large, and therefore liable to considerable steric interactions, the Grignard reagent will want to access the ring from a position where it can deliver the methyl group, easily bond to the carbonyl and avoid any steric clash with the 7 membered ring. This is shown in 5e and rotating the jmol below can help to visualise this.
These steric considerations coupled with the fact that there are many low energy conformers with the carbonyl pointing above the ring cause the methyl to be added from above the plane of the ring as it is dawn here.
Amide Derivative
In an analogous reaction with a prolinol derivative and aniline amination occurs at the 4 position of the pyridinium ring with the same stereochemical outcome.
This phenomenon was investigated in exactly the same way as above. Again the energy of the reactant was minimised using the MM2 method and the figure below shows the conformer of minimum energy with the amide oxygen equatorial and pointing upward.
-
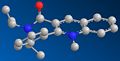 7a. Equatorial (15.9337kcal/mol)
7a. Equatorial (15.9337kcal/mol) -
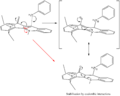 7b. Formation of Stabilised Amine
7b. Formation of Stabilised Amine


In this case the preferred conformer of the reactant determine the stereochemistry of the product. The amine will attach on the same face as the carbonyl since the resulting tertiary amine centred cation is stabilised by resonance enhanced coulombic interactions between itself and the carbonyl oxygen. This is shown in the graphic 7b..
Stereochemistry and Reactivity of an Intermediate in the Synthesis of Taxol
To start with the two intermediates were drawn in ChemDraw, pasted into Chem3D and optimised. Taxol 10, with the carbonyl pointing downward, gave the result shown first in the table below. Taxol 11 gave a result with the carbonyl planar, Taxol 11. This result was of much higher energy. In order to achieve a downward pointing carbonyl the oxygen was manually edited and some of the cyclohexane atoms were moved up above the ring, thus giving Taxol 11 1st Optimisation. Please note that the Jmol buttons are included below to enable the reader to open the molecules in a separate window and orientate them themselves if the graphics are not illustrative enough.
 54.38 kcal/mol 54.38 kcal/mol
|
 124.68 kcal/mol 124.68 kcal/mol
|
 48.15 kcal/mol 48.15 kcal/mol
|
|
|
|
|
These results suggest that 11, with the carbonyl pointing downward is the most stable isomer, however it was believed that the models could be improved. Concentrating on Taxol 10 to start with, the cyclohexane ring seemed to be in a rather strained boat type conformer. The positions of the atoms were manually edited to create a chair type arrangement. After several different arrangements and minimisations a result of lowest energy was found; Taxol 10 optimised Fully.
 41.84 kcal/mol 41.84 kcal/mol
|
 44.31 kcal/mol 44.31 kcal/mol
|
|
|
|
The structures were then examined for any less obvious features that could be optimised.
How One Might Induce Room Temperature Hydrolysis of a Peptide
Axial and equatorial isomers of 13 and 14 were drawn in ChemDraw then copied to ChemDraw3D where their energies were minimised. The results of the minimisation are recorded below, in each case the molecule has been drawn in a chair-chair conformer.
|
|
|
|
|
| 16.426 kcal/mol | 25.8451 kcal/mol | 9.0813 kcal/mol | 14.0496 kcal/mol |
On the basis of this evidence two things are evident, firstly that in both cases the eqautpial conformer is prefered over the axial and secondly the equatorial conformer of 13 is higher in energy than that of 14. (N.B. hydrogenbonding betwen the alohcolic proton and the carbonyl group can possibly be noticed in the eqauorial conformer of 14.)
In order to react with the carbonyl the OH must attack the C=O σ* orbital at the Bürgi-Dunitz angle of 109o. In the 13 equatorial conformer rotation of the N-Cα bond allows facile access to the antibonding orbital and although this may not be the conformer of lowest energy it allows the reaction to take place in accordance with the Curtin-Hammet principle.
In 14 the Cutin-Hammet principle must be invoked even more strongly since in the prefered equatorial conformer despite free rotation about the N-Cα bond the hydroxide cannot easily attain suitable overlap. In order to achieve a suibatle conformeation the hydoxide ring must first flip into the axial conformer in oder to achieve an appropriate orientation.
This explains why the half life of 13 is much shorter than 14. In the later enrgy must be found to convert reactants from the eqautorial to axial conformer, either the enthalpic 5kcal/mol as sugetsed by the result or some other entropic energy, and at room temperature this limits the rate of conversion and so the rate of reaction.
The literature supports this explaination but since I believed that in the axial conformeation the hydroxide and the carboyl were too close together to attain the correct orientation for overlap, a further calculation was carried out. The MM2 minimised axial conformation of 14 was furtther minimised by the B3LYP method and with a 6-31G(D) basis set. The LUMO and HOMO are shown below.

|

|
| LUMO | HOMO |
Unfortunately since the method requires the removal of the lines ddescribing the lone pairs in the gjf input file the HOMO is not centred on the hydroxide oxygen as expected and thus the HOMO and LUMO could not be examined as expected. That said the LUMO is clearly centred on the carbonyl oxygen and it is now much easier to see that with very little rotation about the C-C bond joining the 2 rings in the decalin, the lone pair and the LUMO could overlap thus allowing hydrolysis of the amide bond.
DOI: 10.1021/jo800706y
Modelling Using Semi-empirical Molecular Orbital Theory
Dialkene 12

The dialkene was drawn in ChemDraw and copied into ChemDraw3D. Its energy was minimised using the MM2 method to give a molecule with an energy of 17.9079 kcal/mol. The energy was then minimised to -548182.95 kcal/mol or -873.585857 H and the molecular orbitals calculated using a Hartree Fock Method with a STO-3G basis set, the results of which are shown below.

|

|

|

|

|
| HOMO-1 | HOMO | LUMO | LUMO+1 | LUMO+2 |
The molecule was further minimised using Gaussian, the B3LYP method and a 6-31G(d) basis set. This minimisation was used to calculate the vibrational frequencies. The predicted spectrum is shown below along with the vibration that corresponds to Cl-C and the C=C.

|
||
| IR Spectrum | ||
.jpg)
|
.jpg)
|

|
| C=C stretch 1741cm-1 | C=C stretch 1761cm-1 | C=C stretch 773cm-1 |
The C-Cl strech is expected to occur at 600-800cm-1 and indeed it does at 773cm-1. The alkene stretch is expected at around 1620-1680cm-1 they occur at 1741cm-1 and 1761cm-1 as the alkene is part of a ring system and so has a larger force constant and therefore a higher stretching frequency. The stretching frequency of the C=C which is directly under the Cl atom is higher than the other alkene stretch because Cl can donate its lone pairs in to the C=C π bonding orbital and thus strengthen it. The C-Cl stretch shown has been placed at 773cm-1 due to its intensity although severlal other vibrations inn this area exhibit a much less intense C-Cl stretch.
Alkene 12d
The same procedure was used to calculate the molecular orbitals and energy (-548943.01 kcal/mol or -874.797094 H) of the exo hydrogenated 12d.


|

|

|

|

|
| HOMO-1 | HOMO | LUMO | LUMO+1 | LUMO+2 |
The predicted spectrum is shown below along with the vibration that corresponds to Cl-C and the C=C.

|
.jpg)
|

|
| IR Spectrum | C=C stretch 1757cm-1 | C-Cl stretch 783cm-1 |
The C-Cl stretch has been assigned using the same criterion as above. It should be noticed that the C-Cl stretch is 10cm-1 higher than in the dialkene. This is becasue there is more elctron density in the C-Cl bond and so its bond strength is greater. There is more electron density in the bond since the HOMO-1 is not centred on the π bond and so donation of the lone pairs on Cl is less effective (smaller overlap & greater energy difference denotes weaker bonding).
J.G Starck & H. G. Wallace, "Chemistry Data Book", 2nd Editon, 1984
Miniproject
Introduction
Joelle Prunet heads a research group at Ecole Polytechnique, Paris interested in the synthesis of natural products. They have recently reported a synthesis of Hexacyclinic acid, which includes a rather complicated fused ring system. One of the most interesting steps in this synthesis is the Michael addition that proceeded by an unexpected pathway. The synthesis of the Michael acceptor used in this step proceeds via a RCM and the precursor for the RCM is synthesised by an aldol reaction which produced two products, a cis and trans isomer. The more substituted enolate is formed by the use of catalytic amounts of TiCl4. The reaction then proceeds as usual culminating in dehydration of the aldol product to yield an alkene.

If the mechanism is examined it can be seen that the isomers are formed in the step before dehydration step due to rotation about the C-C bond shown.
Although substantial spectral information has been included in the support information for their journal article the 13C NMR has not been analysed at all. Adjacent peaks have been grouped and arbitrarily assigned to the 2 isomers. The initial aim of this project is to confirm the IR spectrum and assign the 13C data.
Method
In order to produce artificial spectra, each isomer of the molecule was drawn in ChemDraw, transferred to ChemDraw3D and its energy minimised using the MM2 method. This molecule was then submitted to the SCAN and its energy minimised at using a MPW1PW91 DFT method and a 6-31G basis set. The method and basis set was chosen on the advice of the lab script since it is particularly efficient at optimising energy for NMR. The maximum number of cycles was set to 25 to avoid the calculation taking too long; meandering about the optimum energy or the method altering the position of the "bonds", since the molecule is reasonably "floppy" and liable to problems such as these.
After the first optimisation the output file flagged an error in each case.
Considering firtstly the cis siomer on inspection of the data file 3 out of 4 criteria for energy minimisation were met; only the maximum displacement field had not reached the required threshold in the 25 steps.
Item Value Threshold Converged? Maximum Force 0.000086 0.000450 YES RMS Force 0.000008 0.000300 YES Maximum Displacement 0.002610 0.001800 NO RMS Displacement 0.000555 0.001200 YES Predicted change in Energy=-4.604546D-08 Optimization stopped. -- Number of steps exceeded, NStep= 25
The optimisation was considered sucessful since although the Maximum displacement term had not reached the pre determined threshold this was only by 0.000810 H.
In the trans case none of the 4 criteria for energy minimisation had been satisfied, and by a wide margin.
Item Value Threshold Converged? Maximum Force 0.000721 0.000450 NO RMS Force 0.000148 0.000300 YES Maximum Displacement 0.165115 0.001800 NO RMS Displacement 0.035250 0.001200 NO Predicted change in Energy=-3.152656D-05 Optimization stopped. -- Number of steps exceeded, NStep= 25
The output file was re-imputed with the maximum number of cycles set to 25 again and the following result was obtained.
Item Value Thershold Converged? Maximum Force 0.000004 0.000450 YES RMS Force 0.000001 0.000300 YES Maximum Displacement 0.009454 0.001800 NO RMS Displacement 0.001692 0.001200 NO Predicted change in Energy=-2.819008D-09 Optimization completed on the basis of negligible forces. -- Stationary point found.
Convergence to an acceptable degree occured this time since the threshold, although not reached, was in all cases acceptble.
Once the files had been otpimised satisfactoraly they were submitted for NMR analysis and the output files viewed in Gaussian to produce the spectra shown below.
13C NMR
-
 Literature 13C NMR Mix of products
Literature 13C NMR Mix of products -
 13C NMR cis-isomer
13C NMR cis-isomer -
 13C NMR trans-isomer
13C NMR trans-isomer



Reference peak in each case TMS mPW1PW91/6-31G(d,p) CDCl3 GIAO
To start we shall examine the literature reference. The literature reports the 13C NMR spectrum as δ -5.0, -4.1, 13.2 & 13.7, 14.3 & 14.3, 15.8 & 16.0, 18.3 & 18.4, 26.0 & 26.0, 49.2 & 52.5, 61.1 & 61.2, 75.5 & 75.6, 115.8, 136.8 & 138.5, 139.2 & 139.4, 143.9 & 145.5,164.9 & 166.5, 200.2 & 205.2.
| Reference (ppm) | Reference (ppm) | Atom Number | cis (ppm) | Atom Number | trans (ppm) |
|---|---|---|---|---|---|
| 200.2 | 205.2 | 5 | 211.0 | 5 | 203.4 |
| 164.9 | 166.5 | 2 | 168.8 | 2 | 173.1 |
| 143.9 | 145.5 | 6 | 159.0 | 6 | 153.5 |
| 139.2 | 139.4 | 12 | 138.0 | 13 | 138.5 |
| 136.8 | 138.5 | 3 | 129.9 | 3 | 128.6 |
| 115.8 | 15 | 113.7 | 16 | 113.5 | |
| 75.5 | 75.6 | 10 | 72.6 | 11 | 73.0 |
| 61.1 | 61.2 | 13 | 61.5 | 14 | 62.0 |
| 49.2 | 52.5 | 8 | 56.8 | 9 | 56.4 |
| 26.0 | 26.0 | 22 | 23.5 | 23 | 23.5 |
| 18.3 | 18.4 | 20 | 23.3 | 21 | 23.2 |
| 15.8 | 16.0 | 21 | 23.2 | 22 | 23.2 |
| 17 | 20.4 | 18 | 20.2 | ||
| 23 | 17.3 | 7 | 15.8 | ||
| 9 | 15.4 | 10 | 11.3 | ||
| 13.2 | 13.7 | 14 | 11.1 | 15 | 11.2 |
| -4.1 | 19 | -4.2 | 20 | -4.3 | |
| -5.0 | 18 | -7.2 | 19 | -7.4 |
This assignment includes only 16 peaks whereas we would expect 18 since the the molecule has 18 carbon atoms. This fact may suggest that some of the pairs of peaks grouped together are in reality peaks in their own right or that some peaks are lost in the base line.
The predicted 13C NMR is quite a good fit to experiemetnal results. It should be noted that teh molecule cotains and ester carbonyl, namely carbon 2. If we apply the shift correcteion -5ppm then we get much closer to the the refernce value.
Most of the carbon peaks correspond well to the literature and the groupings are mostly correct however some of the peaks seem to have been lost in the baseline. With further time the spectra could be assigned fully and different conformations of particualrly the cis isomer (where most of the ambiguitites lie) could be probed in order to improve the correlation.
IR
-
 IR Spectrum Cis Isomer
IR Spectrum Cis Isomer -
 IR Spectrum Trans Isomer
IR Spectrum Trans Isomer


The literature contained an IR spectrum of a mix of the 2 isomers. The stretching frequencies are compared in the following table.
| Reference (cm-1) | Calc cis (cm-1) | Calc trans (cm-1) |
|---|---|---|
| 2966 | 3126 | 3126 |
| 2935 | 3048 | 3047 |
| 2858 | 1729 | 1745 |
| 1725 | 1702 | 1710 |
| 1702 | 1352 | 1454 |
| 1406 | 1325 | 1354 |
| 1381 | 1313 | 1302 |
| 1252 | 1175 | 1262 |
| 1193 | 1095 | 1187 |
| 1134 | 1080 | 1075 |
| 1068 | 1071 | 1065 |
| 1029 | 1060 | 1060 |
It can be seen that the frequencies do not match up exactly but we must remeber that the calculated frequencies are subject to an error of 8% at the higher wavenumbers and thereforethe calculated values are not as bad as they may appear at first glance.
Conclusion
The IR data correlates well with the experimental, apart from the vibration at 1406cm<-1>> however the 13C shows some abiguities, particualrly with the cis isomer. This may be because the energy minimisation did not fully complete. Repetition of the minimisation would almost certainly improve the data.
References
J. Toueg & J. Prunet, Org. Lett., 2008, 10, 1, 45 DOI 10.1021/ol702566c
NMR cis http://hdl.handle.net/10042/to-1772
IR cis http://hdl.handle.net/10042/to-1773
NMR trans http://hdl.handle.net/10042/to-1774
IR trans http://hdl.handle.net/10042/to-1775