
Rep:Mod:ivyzhang
Module 1: Structure and spectroscopy (Molecular mechanics and molecular orbital)
The Hydrogenation of Cyclopentadiene Dimer
Dimerisation of cyclopentadiene
MM2 force field option was carried out to optimise the geometries for product 1and 2:
| Exo dimer1 | Endo dimer2 |
|---|---|
| ------------MM2 Minimization------------
Note All parameters used are finalized (Quality = 4). Iteration 36 Minimization terminated normally because the gradient norm is less than the minimum gradient norm |
------------MM2 Minimization------------
Note All parameters used are finalized (Quality = 4). Iteration 39 Minimization terminated normally because the gradient norm is less than the minimum gradient norm |
Stretch: 1.2923 Bend: 20.5870 Stretch-Bend: -0.8413 Torsion: 7.6715 Non-1,4 VDW: -1.4358 1,4 VDW: 4.2320 Dipole/Dipole: 0.3778 Total Energy: 31.8834 kcal/mol Calculation completed |
Stretch: 1.2454 Bend: 20.8603 Stretch-Bend: -0.8320 Torsion: 9.5039 Non-1,4 VDW: -1.5083 1,4 VDW: 4.3012 Dipole/Dipole: 0.4448 Total Energy: 34.0153 kcal/mol Calculation completed |
As can be seen, the exo dimer 1 is thermodynamically more stable than the endo dimer 2 by 2.1321kcal/mol. However, the dimerisation of cyclopentadiene produce specifically the endo dimer 2 rather than the exo dimer 1 which contradicts with what has been predicted in the MM2 calculation, this is because the MM2 calculation does not take into account the kinetic control which also has an influence on the selectivity of the products formed. In conclusion, the exo dimer 1 forms faster (kinetically controlled product) than the endo dimer 2 because the activation energy for 1 is lower than that for 2, but 2 is the more stable product (thermodynamically controlled product).
(Jmols of endo + exo forms)
Hydrogenation of dicyclopentadiene
MM2 force field option was carried out to optimise the geometries for product 1and 2
| Dihydro derivative 3 | Dihydro derivative 4 |
|---|---|
| ------------MM2 Minimization------------
Note All parameters used are finalized (Quality = 4). Iteration 36 Minimization terminated normally because the gradient norm is less than the minimum gradient norm |
------------MM2 Minimization------------
Note All parameters used are finalized (Quality = 4). Iteration 59 Minimization terminated normally because the gradient norm is less than the minimum gradient norm |
Stretch: 1.2324 Bend: 18.8641 Stretch-Bend: -0.7625 Torsion: 12.2469 Non-1,4 VDW: -1.5627 1,4 VDW: 5.7523 Dipole/Dipole: 0.1631 Total Energy: 35.9337 kcal/mol Calculation completed |
Stretch: 1.0963 Bend: 14.5074 Stretch-Bend: -0.5493 Torsion: Non-1,4 VDW: -1.0507 1,4 VDW: 4.5124 Dipole/Dipole: 0.1407 Total Energy: 31.1540 kcal/mol Calculation 12.4972completed |
From the MM2 calculation obtained above, the dihydro derivative 4 is 4.7797kcal/mol lower in energy therefore more stable than dihydro derivative 3 in the thermodynamic sense.
This difference in energy is contributed by:
| Dihydro derivative3 | Dihydro derivative4 | Energy difference (3-4) | Contribution/% | |
|---|---|---|---|---|
| stretching (str) | bending (bnd) | torsion (tor) | van der Waals (vdw) | hydrogen bonding (H-Bond) |
| 1.2324 | 18.8641 | 12.2469 | 5.7523 | 0.1631 |
| 1.0963 | 14.5074 | 12.4972 | 4.5124 | 0.1407 |
| 0.1361 | 4.3567 | -0.2503 | 1.2399 | 0.0224 |
| 2.8 | 91.2 | -5.2 | 25.9 | 0.5 |
The bending contributes predominately to this stabilisation which is consistence with the Jmols for 3 and 4, as the structure for 3 looks more bended, hence more strained and/or hindered thermodynamically. The other main contribution to this stabilisation is the van der Waals interaction (25.9 %.) It is also interest to note the torsion strain is actually less for product 3 than 4, however it is a minor effect.
As the MM2 calculation optimises the geometries thermodynamically, it is not enough for us to conclude which hydrogenation product would form as the kinetic control is needed to be considered in order to tell the whole story.
-
 Exo dimer1
Exo dimer1 -
 Endo dimer2
Endo dimer2 -
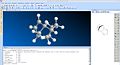 Dihydro derivative3
Dihydro derivative3 -
 Dihydro derivative4
Dihydro derivative4




Click to view the Jmols for 4 different molecules:
|
|
|
|
|
Stereochemistry of Nucleophilic additions to a pyridinium ring (NAD+ analogue)
Reaction mechanism 1

| MM2 | Comment | |
|---|---|---|
| Molecule 5 | ------------MM2 Minimization------------
Pi System: 1 2 3 4 6 5 11 12 Warning: Some parameters are guessed (Quality = 1). Iteration 67 Minimization terminated normally because the gradient norm is less than the minimum gradient norm Stretch: 1.1972 Bend: 11.4383 Stretch-Bend: 0.0535 Torsion: 5.0007 Non-1,4 VDW: -1.9844 1,4 VDW: 11.9030 Charge/Dipole: 2.7063 Dipole/Dipole: -3.9666 Total Energy: 26.3481 kcal/mol Calculation completed MM2 Minimization------------ Separating coincident atoms: Lp Lp [Lp(32)-Lp(33)] Pi System: 1 2 3 4 6 5 11 12 Warning: Some parameters are guessed (Quality = 1). Iteration 152 Minimization terminated normally because the gradient norm is less than the minimum gradient norm Stretch: 0.8899 Bend: 6.8818 Stretch-Bend: 0.1251 Torsion: 9.5042 Non-1,4 VDW: -2.6895 1,4 VDW: 11.5049 Charge/Dipole: 3.1792 Dipole/Dipole: -3.8111 Total Energy: 25.5845 kcal/mol Calculation completed |
MM2 force field option was carried out to optimise the geometries for molecule 5.
The total energy obtained is 26.3481 kcal/mol.  The spatial arrangement of C10, O11, N9, C8, C7, O6 were altered manually and MM2 was run again, the total energy was minimized to 25.5845 kcal/mol with dihedral angle O(11)-C(10)-C(4)-C(3)= -141.3053̊. Click here to see the Jmol of optimised molecule 5
 |
| Molecule 6 | ------------MM2 Minimization------------
Pi System: 1 2 3 4 5 11 12 Warning: Some parameters are guessed (Quality = 1). Iteration 108: Minimization terminated normally because the gradient norm is less than the minimum gradient norm Stretch: 1.4711 Bend: 14.6296 Stretch-Bend: 0.1653 Torsion: 5.4479 Non-1,4 VDW: -2.0418 1,4 VDW: 13.2634 Dipole/Dipole: -4.0662 Total Energy: 28.8694 kcal/mol Calculation completed MM2 Minimization------------ Pi System: 1 2 3 4 5 11 12 Warning: Some parameters are guessed (Quality = 1). Iteration 98: Minimization terminated normally because the gradient norm is less than the minimum gradient norm Stretch: 0.9749 Bend: 9.4470 Stretch-Bend: 0.2180 Torsion: 11.2599 Non-1,4 VDW: -2.6017 1,4 VDW: 12.9210 Dipole/Dipole: -3.9204 Total Energy: 28.2986 kcal/mol Calculation completed |
MM2 force field option was carried out to optimise the geometries for molecule 5.
The total energy obtained is 28.8694kcal/mol.  The spatial arrangement of C12, O13, N11, C10, C9, O8 were altered manually and MM2 was run again, the total energy was minimized to 28.2986 kcal/mol with dihedral angle C(4)-C(5)-C(11)-O(12)= -145.7552̊ . Click here to see the Jmol of optimised molecule 6 The dihedral angle of optimised molecule 6  |
The nucleophilic addition of Grignard to molecule 5 is highly region- and stereoselective. This is because of the chelating effect between the carbonyl oxygen and the magnesium of Grignard reagent leads to a six-membered transition state and thus the methyl anion can only attack from the same face of the carbonyl group and give rise to the optimised geometry in molecule 61. As can be seen, the optimised total energy for molecule 5 is 2.7141kcal/mol less than that for molecule 6, which means the molecule 6 is not the thermodynamically favoured product; therefore this reaction is under kinetic control
Reaction mechanism 2

| MM2 | Comment | |
|---|---|---|
| Molecule 7 | ------------MM2 Minimization------------
Pi System: 21 12 11 17 16 6 15 1 18 2 3 5 4 20 7 8 22 23 Warning: Some parameters are guessed (Quality = 1). Iteration 359: Minimization terminated in error because of repeated or severe errors from the line search Stretch: 2.5513 Bend: 9.7253 Stretch-Bend: 0.3651 Torsion: 5.7329 Non-1,4 VDW: 0.7478 1,4 VDW: 17.4250 Charge/Dipole: 0.6827 Dipole/Dipole: -4.8148 Total Energy: 32.4152 kcal/mol Calculation completed 11,10,9,13,8,24 MM2 Minimization------------ Pi System: 21 12 11 17 16 6 15 1 18 2 3 5 4 20 7 8 22 23 Warning: Some parameters are guessed (Quality = 1). Iteration 2: Minimization terminated normally because the gradient norm is less than the minimum gradient norm Stretch: 1.6571 Bend: 7.7525 Stretch-Bend: 0.3622 Torsion: -6.6293 Non-1,4 VDW: -1.4915 1,4 VDW: 17.5518 Charge/Dipole: 2.6464 Dipole/Dipole: -4.7803 Total Energy: 17.0687 kcal/mol Calculation completed |
MM2 force field option was carried out to optimise the geometries for molecule 7.
The total energy obtained is 32.4152kcal/mol.  The spatial arrangement of C10, O11, N9, C13, C12, C18, C8, C24 were altered manually and MM2 was run again, the total energy was minimized to 17.0687 kcal/mol with dihedral angle O(11)-C(10)-C(4)-C(3)= -136̊. Click here to see the Jmol of optimised molecule 7 The dihedral angle of optimised molecule 7  |
| Molecule 8 | ------------MM2 Minimization------------
Pi System: 28 27 24 26 25 29 30 Pi System: 21 12 11 17 16 15 1 18 2 3 5 4 20 7 8 22 23 Warning: Some parameters are guessed (Quality = 1). Iteration 260: Minimization terminated in error because of repeated or severe errors from the line search Stretch: 1.3900 Bend: 14.8079 Stretch-Bend: 0.3985 Torsion: -31.6204 Non-1,4 VDW: -6.6012 1,4 VDW: 14.2096 Dipole/Dipole: -6.3135 Total Energy: -13.7292 kcal/mol Calculation completed MM2 Minimization------------ Pi System: 28 27 24 26 25 29 30 Pi System: 21 12 11 17 16 15 1 18 2 3 5 4 20 7 8 22 23 Warning: Some parameters are guessed (Quality = 1). Iteration 152: Minimization terminated in error because of repeated or severe errors from the line search Stretch: 1.2479 Bend: 14.5280 Stretch-Bend: 0.2876 Torsion: -30.6165 Non-1,4 VDW: -7.8148 1,4 VDW: 14.2034 Dipole/Dipole: -6.6411 Total Energy: -14.8055 kcal/mol Calculation completed |
MM2 force field option was carried out to optimise the geometries for molecule 8.
The total energy obtained is -13.7292 kcal/mol.  The spatial arrangement of C12, O13, N11, C10, C9, O8 were altered manually and MM2 was run again, the total energy was minimized to -14.8055 kcal/mol with dihedral angle C(4)-C(5)-C(10)-O(11)= -134̊ . Click here to see the Jmol of optimised molecule 8 The dihedral angle of optimised molecule 8  |
The nucleophilic addition of PhNH2 is highly diastereoselective and governed by the chiral axis C3-C=O. Diffeent from reaction 1, the nucleophile in this case attacks from the opposite face of the carbonyl group orientation as a result of the stereohinderance between the very bulky PhNH2 and carbonyl group is minimise2. The molecule 7 is much higher in energy than molecule 8, which means the product 8 is thermodynamically favoured.
Stereochemistry and Reactivity of an Intermediate in the Synthesis of Taxol
| MM2 | Comment | |
|---|---|---|
| Molecule 10 | ------------MM2 Minimization------------
Note: All parameters used are finalized (Quality = 4). Iteration 124: Minimization terminated normally because the gradient norm is less than the minimum gradient norm Stretch: 2.5362 Bend: 11.7205 Stretch-Bend: 0.3914 Torsion: 19.0131 Non-1,4 VDW: -0.8866 1,4 VDW: 12.4409 Dipole/Dipole: 0.1438 Total Energy: 45.3594 kcal/mol Calculation completed MM2 Minimization------------ Note: All parameters used are finalized (Quality = 4). Iteration 226: Minimization terminated normally because the gradient norm is less than the minimum gradient norm Stretch: 2.5563 Bend: 10.6458 Stretch-Bend: 0.3241 Torsion: 19.6527 Non-1,4 VDW: -1.2454 1,4 VDW: 12.5391 Dipole/Dipole: -0.1814 Total Energy: 44.2912 kcal/mol Calculation completed |
MM2 force field option was carried out to optimise the geometries for molecule 10.
The total energy obtained is: 45.3594 kcal/mol.  The spatial arrangement of C12, O14, C8, C2,C3 were altered manually so that the carbon ring obtains a twisted boat conformation with the C=O orientated in the same direction as the methylene bridge3, MM2 was run again, the total energy was minimized to 44.2912 kcal/mol kcal/mol. Click here to see the Jmol of optimised molecule 10
|
| Molecule 11 | ------------MM2 Minimization------------
Note: All parameters used are finalized (Quality = 4). Iteration 124: Minimization terminated normally because the gradient norm is less than the minimum gradient norm Stretch: 2.5419 Bend: 11.6952 Stretch-Bend: 0.3875 Torsion: 19.0276 Non-1,4 VDW: -0.8603 1,4 VDW: 12.4287 Dipole/Dipole: 0.1447 Total Energy: 45.3653 kcal/mol Calculation completed MM2 Minimization------------ Note: All parameters used are finalized (Quality = 4). Iteration 219: Minimization terminated normally because the gradient norm is less than the minimum gradient norm Stretch: 2.5729 Bend: 9.9777 Stretch-Bend: 0.2795 Torsion: 19.3062 Non-1,4 VDW: -1.4586 1,4 VDW: 12.6542 Dipole/Dipole: -0.1873 Total Energy: 43.1445 kcal/mol Calculation completed |
MM2 force field option was carried out to optimise the geometries for molecule 11.
The total energy obtained is 45.3653 kcal/mol  The spatial arrangement of C3, O17, C12, C8, C7, O2 were altered manually so that the carbon ring obtains a chair conformation with the C=O orientated in the opposite direction as the methylene bridge, the total energy was minimized 43.1445 kcal/mol. Click here to see the Jmol of optimised molecule 11 |
Both isomers have similar energies; isomer 10 is slight higher in energy than isomer 11 thus the later one will form the more stable intermediate in the carbonyl addition reaction. For the isomer 10, it adopts a thermodynamically unfavoured twisted-boat conformation as the positioned E-cyclononenone bridge locks the fused carbon ring due to its rigidity. Whereas for isomer 11, the nonbonded transannular interactions within the core ring make the chair conformation more favourable3.
Why the alkene reacts slowly?
As the alkene is adjacent to a bridgedhead, it reacts more slowly as it is thermodynamically more stable due to the hyperconjugation between the π anti-bonding and C-H bonding orbital.
How one might induce room temperature hydrolysis of a peptide
Reaction 1

Reation 2

Decalin exists in trans (with OH group axial) and cis (with OH group equatorial) forms. The energetically stable form(less steric interactions) will act as the major reactant.
| Diastereoisomers of cis-decalin | MM2 | Comment |
|---|---|---|
Axial carboxamide
 |
------------MM2 Minimization------------
Warning: Some parameters are guessed (Quality = 1). Iteration 19: Minimization terminated normally because the gradient norm is less than the minimum gradient norm Stretch: 1.6948 Bend: 8.4913 Stretch-Bend: 0.6464 Torsion: 11.9016 Non-1,4 VDW: -7.1847 1,4 VDW: 10.3567 Dipole/Dipole: -3.6258 Total Energy: 22.2803 kcal/mol Calculation completed
|
From the MM2 calculation, both diastereoisomers obtain the chair conformation with the axial isomer 5.7776kcal/mol higher in energy than that of the equatorial isomer. This result is well matched with the literature energy difference (5.0 kcal/mol) and hence the equatorial isomer is the major form of the cis-decalin. The reason behinds this energy difference is that the equatorial ethylamido is stabilised by the intra-molecular interaction between the oxygen lone pair on the hydroxyl group and the hydrogen on the amine, whereas the axial ethylamido is destabilised by the syn-diaxial interactions4.
Click here to view the Jmol of optimised axial cis-decline |
Equatorial carboxamide
 |
------------MM2 Minimization------------
Warning: Some parameters are guessed (Quality = 1). Iteration 26: Minimization terminated normally because the gradient norm is less than the minimum gradient norm Stretch: 1.6703 Bend: 3.6616 Stretch-Bend: 0.5234 Torsion: 7.6515 Non-1,4 VDW: -3.3846 1,4 VDW: 9.6401 Dipole/Dipole: -3.2596 Total Energy: 16.5027 kcal/mol Calculation completed |
Click here to view the Jmol of optimised equatorial cis-decline
|
| Diastereoisomers of trans-decalin | MM2 | Comment |
|---|---|---|
Axial carboxamide
 |
------------MM2 Minimization------------
Warning: Some parameters are guessed (Quality = 1). Iteration 23: Minimization terminated normally because the gradient norm is less than the minimum gradient norm Stretch: 1.5110 Bend: 4.0925 Stretch-Bend: 0.5184 Torsion: 7.3740 Non-1,4 VDW: -6.8650 1,4 VDW: 9.9111 Dipole/Dipole: -4.5255 Total Energy: 12.0165 kcal/mol Calculation completed
|
From the MM2 calculation, both diastereoisomers obtain the chair conformation with the axial isomer 2.9697kcal/mol higher in energy than that of the equatorial isomer. This result is well matched with the literature energy difference (3.1kcal/mol) and hence the equatorial isomer is the major form of the trans-decalin. The reason behinds this energy difference is the same as that for the cis-diastereoisomer 4.
Click here to view the Jmol of optimised axial trans-decline |
Equatorial carboxamide
 |
------------MM2 Minimization------------
Warning: Some parameters are guessed (Quality = 1). Iteration 22: Minimization terminated normally because the gradient norm is less than the minimum gradient norm Stretch: 1.5363 Bend: 3.2669 Stretch-Bend: 0.4873 Torsion: 7.2203 Non-1,4 VDW: -8.1271 1,4 VDW: 9.7583 Dipole/Dipole: -5.0952 Total Energy: 9.0468 kcal/mol Calculation completed |
Click here to view the Jmol of optimised equitorial trans-decline
|
The hydrolysis for the cis-decalin happens much faster than the trans-decalin due to the reasons below:
The equatorial ethylamido group in the cis-decalin undergoes intramolecular nucleophilic attack by the hydroxyl group in the correct orientation. Whereas the equatorial ethylamido group in the trans-decalin must invert to the axial conformation in order for this reaction to happen4.
Reaction scheme for cis-decalin

Reaction scheme for trans-decalin

Modelling Using Semi-empirical Molecular Orbital Theory
| MM2 | Comment | |
|---|---|---|
| Molecule 12 | ------------MM2 Minimization------------
Warning: Some parameters are guessed (Quality = 1). Iteration 116: Minimization terminated normally because the gradient norm is less than the minimum gradient norm Stretch: 0.6294 Bend: 4.6483 Stretch-Bend: 0.0388 Torsion: 7.7307 Non-1,4 VDW: -1.0746 1,4 VDW: 5.8176 Dipole/Dipole: 0.1128 Total Energy: 17.9031 kcal/mol Calculation completed |
MM2 force field option was carried out to optimise the geometries for molecule 12, the optimized geometry was obtained in boat conformation.
The total energy obtained is 17.9031 kcal/mol.  Click here to view the Jmol of optimised molecule 12 |
| Molecule 13 | ------------MM2 Minimization------------
Warning: Some parameters are guessed (Quality = 1). Iteration 80: Minimization terminated normally because the gradient norm is less than the minimum gradient norm Stretch: 0.8962 Bend: 4.6688 Stretch-Bend: 0.0120 Torsion: 10.8017 Non-1,4 VDW: -1.1116 1,4 VDW: 7.0083 Dipole/Dipole: 0.0705 Total Energy: 22.3458 kcal/mol Calculation completed |
MM2 force field option was carried out to optimise the geometries for molecule 13.
The total energy obtained is 22.3458 kcal/mol.  Click here to see the Jmol of optimised molecule 13 |
| HF/STO-3G | Comment | |
|---|---|---|
| Molecule 12 | ----------- Gaussian Interface ------------
Model: 5.mol 1) Gaussian Job: # RHF/STO-3G Opt Test Finished @ Energy = -548182.95 Kcal/Mol (-873.585857 Hartrees) |
The HF/STO-3G self-consistent-field MO method was carried out to provide an approximate representation of the valence-electron molecular wavefunction, no obvious change in the optimized geometry due to the rigidity of the molecule.
Click here to view the Jmol of optimised molecule 12 |
| Molecule 13 | ------------ Gaussian Interface ------------
Model: 5.mol 1) Gaussian Job: # RHF/STO-3G Opt Test Finished @ Energy = -548945.59 Kcal/Mol (-874.801205 Hartrees)
|
The HF/STO-3G self-consistent-field MO method was carried out to provide an approximate representation of the valence-electron molecular wavefunction, no obvious change in the optimized geometry due to the rigidity of the molecule.
Click here to see the Jmol of optimised molecule 13 |
| Orbital name | Images(isocontour=0.02) | Comment5 |
|---|---|---|
| HOMO-1 |  |
Exo π C=C |
| HOMO |  |
Endo σ C=C* |
| LUMO |  |
/ |
| LUMO+1 |  |
/ |
| LUMO+2 |  |
σ C-Cl* |
The optimized gjf files were send to SCAN and IR spectra are analyzed as below:
| Assignment | Wavenumber/cm-1 | Intensity | |
|---|---|---|---|
Molecule 12
 |
C-Cl stretch Exo C=C stretch Endo C=C stretch |
770.951 1737.20 1757.39 |
25.1237 4.212 3.927 |
| Assignment | Wavenumber/cm-1 | Intensity | |
|---|---|---|---|
Molecule 13
 |
C-Cl stretch Endo C=C stretch |
774.967 1758.07 |
19.9619 4.3511 |
The C-Cl stretching frequency is larger for molecule 13 than that for molecule 12 due to the exo π C=C orbital donates its electron density into the σ C-Cl* orbital which weakens the bond strength but increased the bond length5.
Mini Project: Total synthesis of aspergillide B and structural discrepancy of aspergillide A
Introduction:
Aspergillides A and B can be obtained via the Pd (II)-catalyzed stereospecific synthesis of tetrahydropyrans which we developed for the synthesis of some natural products.
Structure of A and B:

Click here to see the Jmol of optimised A
Click here to see the Jmol of optimised B
The Retrosynthetic Analysis for Aspergillides:

Results and Discussion: MM2 was used to optimise the geometris of A and B initially, followed by the HF/STO-3G. The obtained gjf files were then send to SCAN. The NMR, IR and Optical Rotation results are listed and analyzed as beblow:
-
 NMR results
NMR results -
 IR results
IR results -
 Optical Rotation results
Optical Rotation results
For NMR, each calculated chemical shift has its corrsponding literature value and the difference between them are reasonably small. Therefore, GIAO can be used as an appropriate method to obtain the NMR data for the Aspergillides. For IR, the literature and the calculated data are fairly close but for some absorptions, the calculated frequency does not give a good enough indication, this might due to the stretching and bending interactions which are very close together, therefore interference one another, as a result, the calcualted IR is less close to the experimental data. For optical rotation, the calculated and literature values are so different, however they gives the same sigh which means it can be only used to predict whether the product is R- or S- stereoisomer and no useful information on the actual optical rotation value.
References:
1. A. G. Shultz, L. Flood and J. P. Springer, J. Org. Chemistry, 1986, 51, 838. DOI:10.1021/jo00356a016
2. Leleu, Stephane; Papamicael, Cyril; Marsais, Francis; Dupas, Georges; Levacher, Vincent. Tetrahedron: Asymmetry, 2004, 15, 3919-3928. DOI:10.1016/j.tetasy.2004.11.004
3. S. W. Elmore and L. Paquette, Tetrahedron Letters, 1991, 319; DOI:10.1016/S0040-4039(00)92617-0 10.1016/S0040-4039(00)92617-0 10.1016/S0040-4039(00)92617-0
4. M. Fernandes, F. Fache, M. Rosen, P.-L. Nguyen, and D. E. Hansen, 'Rapid Cleavage of Unactivated, Unstrained Amide Bonds at Neutral pH', J. Org. Chem., 2008, 73, 6413–6416 ASAP: DOI:10.1021/jo800706y
5. B. Halton, R. Boese and H. S. Rzepa., J. Chem. Soc., Perkin Trans 2, 1992, 447. DOI:10.1039/P29920000447
6. Total synthesis of aspergillide B and structural discrepancy of aspergillide A; Sudhir M. Hande, Jun’ichi Uenishi *Kyoto Pharmaceutical University, Misasagi, Yamashina, Kyoto 607-8412, Japan