
Rep:Mod:inorgcomplabdgp12
Week 1: The ropes
Geometry analysis and Optimisations
| BH3 | BBr3 | GaBr3 | |
|---|---|---|---|
| r(E-X)(Å) | 1.192 | 1.93 | 2.35 |
| θ(X-E-X)(°) | 120 | 120 | 120 |
| Summary Data | Convergence | Jmol | |||
|---|---|---|---|---|---|
 |
 |
|
The optimisation file is liked to here for BH3/3-21G
| Summary Data | Convergence | Jmol | |||
|---|---|---|---|---|---|
 |
 |
|
The optimisation file is liked to here for BH3/6-31G
| Summary Data | Convergence | Jmol | |||
|---|---|---|---|---|---|
 |
 |
|
The optimisation file is liked to here GaBr3
| Summary Data | Convergence | Jmol | |||
|---|---|---|---|---|---|
 |
 |
|
The optimisation file is liked to here
Varying the ligands has little to no effect on the bond angles. In each case of BH3, GaBr3 and BBr3 the molecules adopt a trigonal planar structure belonging to the D3h point group. As result in each case the bonds are at 120°.
However, changing the ligand and central atom does have an effect on the bond lengths[1]. When changing the ligand atom H to a Br atom and an increase in bond length is observed due to the larger size of the Br atom. The same aplies when the central atom is changed from boron to gallium. Since gallium is the larger atom an increase in the bond length can observed when comparing GaBr3 to BBr3.
A bond[2] can be described as the electrostatic attraction between two nuclei derived from the overlap of atomic orbitals. There are various types of bond that can be formed, such as ionic, covalent and hydrogen bonds. Ionic and covalent bonds are usually considered to be the stronger form of bond, these are brought about by the sharing of an electron pair between nuclei. Whereas hydrogen bonding or dipole-dipole interactions are considered to be weaker since those rely on electrostatic force based on the distortions of the electron cloud.
An article by R.T. Sanderson [3] will be used to to relatively describe a strong, medium and weak bond. Although generally the strength of a bond is determined by how much energy is needed to break it. In any case, accordance to the article the F-F bond has one of the highest energies at 113.1kcal/mol whereas one of the lowest energy bonds would be Hg-Hg which is quoted at 8.6kcal/mol. A medium bond could be for example Al-Al which has a bond energy of 48.2kcal/mol. If this were applied to the three molecules that were examined the weakest bond would belong to BH3(-1.67x104kcal/mol), then GaBr3 (-2.62x104kcal/mol) and lastly BBr3 (-4.04x104kcal/mol). It is important to note that different basis sets were used to optimise each of the molecules. For the sake of the exercise it is assumed that each basis set predicts the energy to the same extent of accuracy to make the comparison valid.
BH3: B3LYP/6-31G(d,p)
| Summary Data | Frequency |
|---|---|
 |
The frequency output file is liked to here
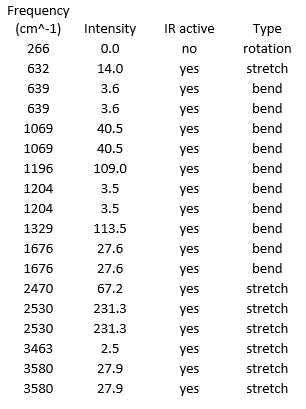
| Wavenumber (cm-1) | Intensity | IR active | Type |
|---|---|---|---|
| 1163 | 93 | yes | bend |
| 1213 | 14 | slightly | bend |
| 1213 | 14 | slightly | bend |
| 2583 | 0 | no | stretch |
| 2716 | 126 | yes | stretch |
| 2716 | 126 | yes | stretch |
A molecular orbital diagram of BH3 is linked here The MOs generated by gaussview are very similar compared to the LCAOs as shown in the link above. As a result qualitative MO analysis provides a fairly detailed model in order to image orbitals.
IR Spectrum of BH3

In the IR spectrum some of the vibrations quoted in the table are not observed due to that particular vibrational having a centre of symmetry and as a result will not be IR active. Additionally there are degenerate vibrations which causes an overlap of peaks on the spectrum.
GaBr3: B3LYP/6-31G(d,p)
| Summary Data | Frequency |
|---|---|
 |
The frequency file is liked to here
| Wavenumber (cm-1) | Intensity | IR active | Type |
|---|---|---|---|
| 76 | 3 | slightly | bend |
| 76 | 3 | slightly | bend |
| 100 | 9 | yes | bend |
| 197 | 0 | no | stretch |
| 316 | 57 | yes | stretch |
| 316 | 57 | yes | stretch |
IR Spectrum of GaBr3

By comparing the frequency analysis of GaBr3 and BH3 a large difference in frequencies can be observed. This can be attributed to the large difference in the weight of the atoms. This results in more work being done in moving the atoms and as a result they will vibrate at a low frequency. This can be illustrated by Hooke's Law: "F=-kx". It can be said the GaBr3 has a higher force constant than BH3 hence more force is required by the gallium tribromide to move the same distance as the boron trihydride.
The third vibrational mode corresponds to the umbrella motion in GaBr3 whereas it is the first mode in BH3. However by analysing the displacement vectors of the two compounds it can be seen that the source of the vibrational motion is different. In gallium tribromide the vibrational motion is largely driven by the displacement of the gallium atom whereas in boron trihydride the vibration is driven by the displacement of the hydrogen atoms.
It is important to use the same basis set for these calculations because it is a reflection of the accuracy that was used to carry out the calculation. More accurate basis sets use additional functions to carry out the same calculation to varying degrees of accuracy hence it is invalid to compare results from different basis sets since one calculation will always have been carried out to a greater degree of accuracy.
The importance of frequency analysis is to find the curvature of the minimum which was found from running the optimisation, and it can be used to confirm the presence of a minimum since the curvature would be positive at that point. Additionally the frequency analysis provides low frequencies which correspond to the molecule's fundemental modes of vibration.
NH3 Optimisation and frequency
| Summary Data | Convergence | Jmol | |||
|---|---|---|---|---|---|
 |
 |
|
The log file is linked here
| Summary Data | Frequency |
|---|---|
 |
The frequency output file is linked here
| Wavenumber (cm-1) | Intensity | IR active | Type |
|---|---|---|---|
| 1098 | 145 | yes | bend |
| 1694 | 14 | yes | bend |
| 1694 | 14 | yes | bend |
| 3461 | 1 | no | stretch |
| 3590 | 0 | no | stretch |
| 3590 | 0 | no | stretch |

NH3BH3
| Summary Data | Convergence | Jmol | |||
|---|---|---|---|---|---|
 |
 |
|
The optimisation file is linked here
| Summary Data | Frequency |
|---|---|
 |
The frequency output file is linked here

{kind=link}
• E(BH3)=-26.6153235 a.u.
• E(NH3)=-56.5577687 a.u.
• E(NH3BH3)=-83.2246889 a.u.
ΔE=E(NH3BH3)-[E(NH3)+E(BH3)] -83.2246889-(-26.6153235+(-56.5577687))=-0.0515967 a.u. = -135.47kJ/mol = -32.38kcal/mol The N-B bond is very weak compared to the bond energies in the 3 starting molecules. The B-H bond was found to be -2.33x104kJmol-1.
Week 2 Project: Ionic Liquids
[N(CH3)4]+
| Summary Data | Convergence | Jmol | |||
|---|---|---|---|---|---|
4-%2B_summary.PNG) |
4-%2B_converge.PNG) |
|
[N(CH3)4]+ optimisation output file.
The molecule adopts a tetrahedral geometry with the Td point group. The N-C and C-H bond distances are 1.51Å and 1.09Å respectively. The bond angles reported are C-N-C and H-C-H are 109.5° and 110.0° respectively.
| Summary Data | Frequency |
|---|---|
4-%2B_frqsummary.PNG) |
[N(CH3)4]+ frequency output file.
4-%2B_IR.PNG)
{kind=link}
Molecular Orbitals
The following are links to 5 molecular orbitals obtained from this molecule. The most anti-bonding molecule is the first in the list and the most bonding orbital at the bottom. Yellow arrows are used to denoted anti-bonding interactions and cyan arrows represent bonding interactions. Dashed arrows are used for through space interactions.
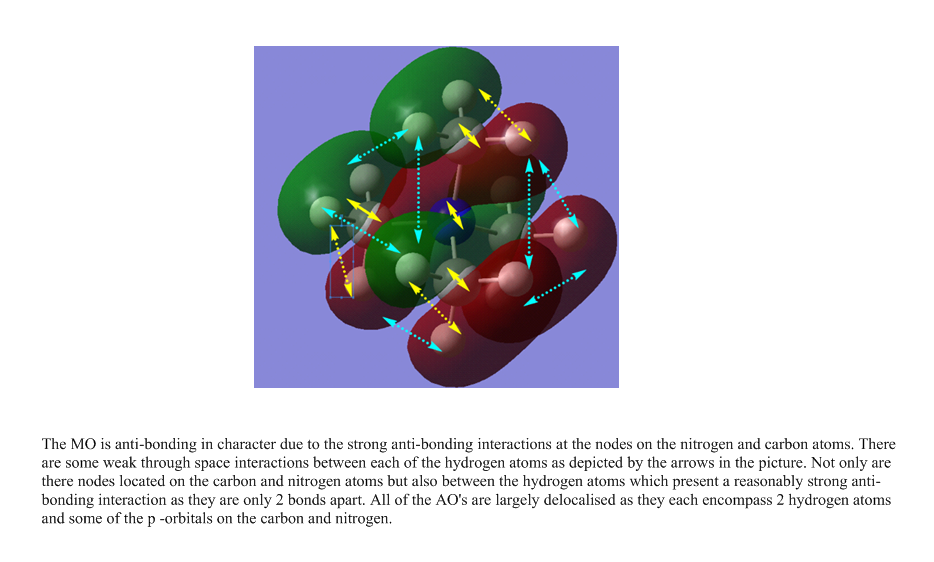{kind=link}
{kind=link}
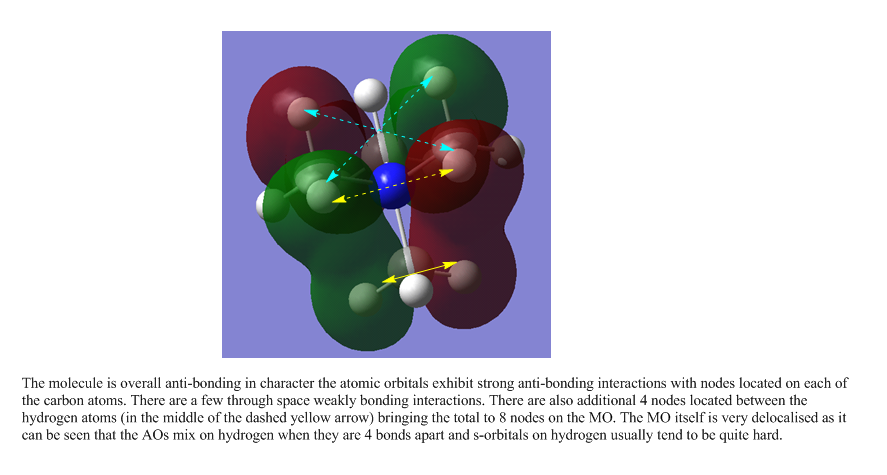{kind=link}
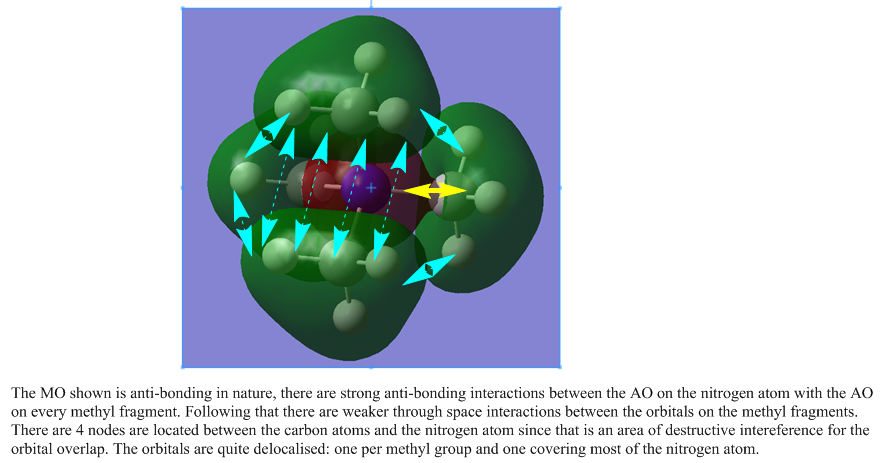{kind=link}
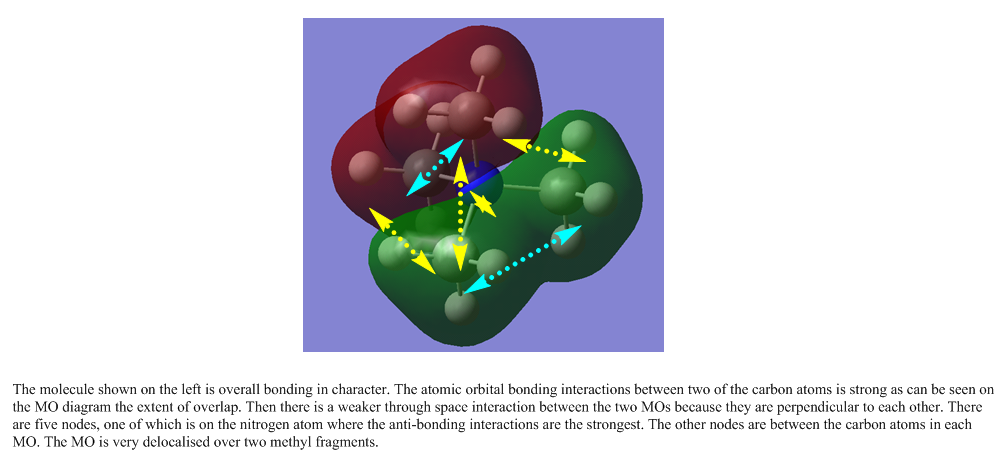{kind=link}
[P(CH3)4]+
| Summary Data | Convergence | Jmol | |||
|---|---|---|---|---|---|
4-%2B_summary.PNG) |
4-%2B_converge.PNG) |
|
[P(CH3)4]+ optimisation output file.
The central geometry around the phosphorus atom is tetrahedral with a Td point group similar to the previous example. The bond lengths for P-C and C-H are 1.82Å and 1.09Å respectively, with bond angles C-P-C, H-C-H being 109.5° and 109.0° respectively. From this it can be determined that the P-C bond length is greater than that for N-C. Nitrogen and phosphorus are both group 5 elements but phosphorus is in the period below which results in it having a greater number of filled orbitals. This results in phosphorus having larger p-orbitals which results in a poorer orbital match with the p-orbital on carbon which results in a longer bond.
| Summary Data | Frequency |
|---|---|
4-%2B_frqsummary.PNG) |
[P(CH3)4]+ frequency output file.
4-%2B_IR.PNG)
{kind=link}
[S(CH3)3]+
| Summary Data | Convergence | Jmol | |||
|---|---|---|---|---|---|
3-%2B_summary.PNG) |
3-%2B_converge.PNG) |
|
[S(CH3)3]+ optimisation output file.
Unlike the previous two cations this one adopts a trigonal pyramidial geometry with a C3V symmetry. The main reason for this change in geometry is brought about by the lone pair on the central atom which was not present in the previous examples. The bonds S-C and C-H 1.82Å and 1.09Å respectively and the angles between C-S-C and H-C-H being 102.7° and 111.1°. The S-C bond is similar in length to the aforementioned P-C bond as there is only one additional electron present which does not have a greater effect. If anything the bond should be slightly shorter because the outer electrons experience a greater Zeff in sulphur causing a reduction in the nuclear radius. The repulsion caused by the lone pair on the S-C bonds could explain this offset since shorter bonds would cause the carbon atoms to be closer which would be electronically and sterically unfavourable.
| Summary Data | Frequency |
|---|---|
3-%2B_frqsummary.PNG) |
[P(CH3)4]+ frequency output file.
3-%2B_IR.PNG)
{kind=link}
NBO analysis
Natural bond order analysis has been carried out for all three molecules: [N(CH3)4]+, [P(CH3)4]+, [S(CH3)4]+. A table illustrating all the charges can be found here. A general trend can be observed, by increasing the electronegativity of the central atom causes the charge on the carbon atom to be reduced. Since the central atom will have a greater affinity for electrons than carbon, less electron density will be deposited onto the carbon.
![[N(CH3)4]+](/images/b/ba/-N%28CH3%294-%2B_coloured_charge.PNG){kind=link}
![[P(CH3)4]+](/images/0/04/-P%28CH3%294-%2B_coloured_charge.PNG){kind=link}
![[S(CH3)4]+](/images/f/f7/-S%28CH3%294-%2B_coloured_charge.PNG){kind=link}
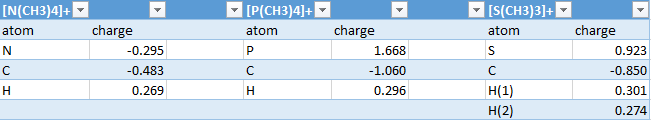{kind=link}
The contribution that the carbon atom makes to the C-X bond where X is the central atom is related to the trend mentioned previously. Carbon contributes the most in the C-P bond whereas it contributes the least in the C-N bond, which corresponds to our earlier statement. There is a greater amount of charge located on carbon when the central atom has lower electronegativity which leaves a larger portion of the electron cloud on carbon which in turn allows carbon to contribute more to the sharing of electrons involved in creating a bond.
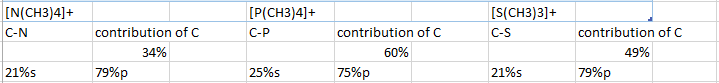{kind=link}
From this study the nitrogen atom in [N(CH3)4]+ is negatively charged. This deviates from traditional structures where a positive charge is placed on it, in accordance with its lewis structure. Nitrogen will need to give up an additional electron to form the 4th bond making it overall positively charged. However, from the NBO analysis it can be seen that the charge is in fact distributed over the hydrogen atoms. It can be said that the molecule forms a shell of positive charge around itself with negatively charged centres.
[N(CH3)3(CH2OH)]+
| Summary Data | Convergence | Jmol | |||
|---|---|---|---|---|---|
3(CH2OH)-%2B_summary.PNG) |
3(CH2OH)-%2B_converge.PNG) |
|
[N(CH3)3(CH2OH)]+ optimisation output file.
| Summary Data | Frequency |
|---|---|
3(CH2OH)-%2B_frqsummary.PNG) |
[N(CH3)3(CH2OH)]+ frequency output file.
3(CH2OH)-%2B_IR.PNG)
{kind=link}
[N(CH3)3(CH2CN)]+
| Summary Data | Convergence | Jmol | |||
|---|---|---|---|---|---|
3(CH2CN)-%2B_summary.PNG) |
3(CH2CN)-%2B_converge.PNG) |
|
[N(CH3)3(CH2CN)]+ optimisation output file.
| Summary Data | Frequency |
|---|---|
3(CH2CN)-%2B_frqsummary.PNG) |
[N(CH3)3(CH2CN)]+ frequency output file.
3(CH2CN)-%2B_IR.PNG)
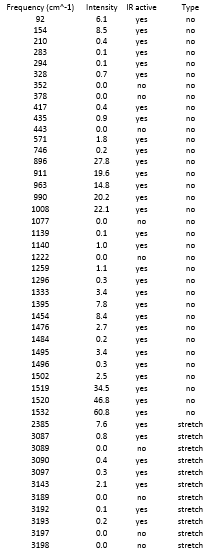{kind=link}
Charge and MOs
The charge distributions on [N(CH3)3(CH2OH)]+ and [N(CH3)3(CH2CN)]+ are summarised in this table. The CN acts as an electron withdrawing group whereas OH acts as an electron donating group in these examples. This is illustrated in the charge distribution when examining the charges on the central atom specifically, the molecule containing the hydroxyl substituent, the charge distributed on the nitrogen atom when compared to [N(CH3)4]+ an increase in negative charge on nitrogen is observed which corresponds to the the electron donating nature of the hydroxyl group. Conversely when using the cyano substituent an increase in positive charge is observed on the central nitrogen atom relative to the unsubstituted component. In all cases the positive charge which make the molecules cationic is spread over the hydrogen atoms.
![[N(CH3)3(CH2OH)]+](/images/1/10/-N%28CH3%293%28CH2OH%29-%2B_coloured_charge.PNG){kind=link}
![[N(CH3)3(CH2CN)]+](/images/4/4e/-N%28CH3%293%28CH2CN%29-%2B_coloured_charge.PNG){kind=link}
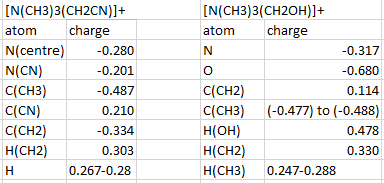{kind=link}
In contrast another aspect worth commenting on is the charge found on the carbon atom in the methylene group, the OH group causes a large increase of positive charge on the carbon atom in the methylene group whereas the CN group causes a lesser increase in positive charge on the methylene carbon relative to [N(CH3)4]+. The increase is expected for CN as it is an electron donating group however it is unexpected for the OH group. One possible explanation would be that the orbitals on oxygen match up better to the orbitals on nitrogen than carbon which causes it to preferentially donate to nitrogen and that additional electron density is withdrawn from the methylene carbon due to the greater electron affinity an oxygen atom holds. This would explain why most of the negative charge is concentrated on the oxygen atom.
A comparison of the HOMOs of [N(CH3)4]+, [N(CH3)3(CH2OH)]+ and [N(CH3)3(CH2CN)]+ showed changes in their energy and shape. In terms of energy relative to [N(CH3)4]+, the molecule with the hydroxyl substituent caused the HOMO to increase in energy whereas, for the cyano substituent a decrease was obsvered which ties in with the previous argument for the charge distributions. The electron withdrawing group (CN) has a greater affinity for electrons, this results in a decrease in energy for the AOs becuase the orbitals become more contracted. In terms of the shape of the electron cloud, it becomes a lot more concentrated around the functional group, and it is no longer spread around the entire molecule as was shown for [N(CH3)4]+.
![[N(CH3)4]+](/images/b/be/-N%28CH3%294-%2B_HOMO.PNG){kind=link}
![[N(CH3)3(CH2OH)]+](/images/8/85/-N%28CH3%293%28CH2-oh%29-%2B_HOMO.PNG){kind=link}
![[N(CH3)3(CH2CN)]+](/images/c/cb/-N%28CH3%293%28CH2CN%29-%2B_HOMO.PNG){kind=link}
In contrast the LUMOs of [N(CH3)4]+, [N(CH3)3(CH2OH)]+ and [N(CH3)3(CH2CN)]+ showed changes in their energy and shape. For the substituted molecules a decrease in the energy of the LUMO is observed. In terms of shape however, the LUMOs are quite similar they all engulf the entire molecule in its electron cloud. Upon examination of the HOMO-LUMO gap in these three molecules it can be seen that the substituted molecules have a smaller gap compared to the unsubstituted cation. Which results in an increase in reactivity for [N(CH3)3(CH2OH)]+ and [N(CH3)3(CH2CN)]+. This change in chemical reacitivity is brought about by the need for less energy in the promotion of electrons accross the HOMO-LUMO gap, hence its susceptibility to chemical reaction has been enhanced.
![[N(CH3)4]+](/images/7/73/-N%28CH3%294-%2B_LUMO.PNG){kind=link}
![[N(CH3)3(CH2OH)]+](/images/1/18/-N%28CH3%293%28CH2-oh%29-%2B_LUMO.PNG){kind=link}
![[N(CH3)3(CH2CN)]+](/images/0/0d/-N%28CH3%293%28CH2CN%29-%2B_LUMO.PNG){kind=link}