
Rep:Mod:hcn06report1
The basic techniques of molecular mechanics and semi-empirical molecular orbital methods for structural and spectroscopic evaluations
Hydrogenation of cyclopentadiene Dimer
Geometries optimised using the Moleuclar Mechanics (MM2) force field option:

An isomer with the substituent (the cyclopentene in this case) located furthest away from the longest bridge, which contains most atoms.

An isomer with the substituent (the cyclopentene in this case) located closest to the longest bridge, which contains most atoms.
By comparing the energies of the two dimers, it can be seen that the endo dimer 2 with higher energy (34.0153 kcal mol-1) is more strained than the exo dimer 1 (31.8834 kcal mol-1) in a thermodynamic sense. However, the endo dimer is observed to be more stable than the exo dimer in the cyclodimerisation of cyclopentadiene due to kinetic (ie transition state stability) factors (the secondary oribital interactions). The stereoselectivity is controlled by the electronic properties of the molecule. The cyclodimerisation of cyclopentadiene occurs between the HOMO of a diene and the LUMO of a dienophile. The carbons with the highest coefficients in the two frontier orbitals will bond. Therefore, they determine the orientation of the substituent and identify the major product.
Geometries optimised using the MM2 force field option:

A dihydro derivative formed by hydrogenation of the double bond on the cyclopentene of the endo dimer 2.

A dihydro derivative formed by hydrogenation of the double bond on the cyclohexene of the endo dimer 2.
By comparing the energies of the two dihydro derivatives, it can be seen that the dihydro derivative 4 with lower energy (31.1540 kcal mol-1) is thermodynamically (ie product stability) favoured over the dihydro derivative 3 (35.9323 kcal mol-1) in the hydrogenation reaction of the dimer and the dihydro derivative 3 is more strained. This also shows that the double bond on the cyclohexene of the endo dimer 2 eases the hydrogenation.
The table below shows the relative contributions of different energy terms of the two dihydro derivatives:
| Energy-terms | Energy for Dihydro derivative 3(kcal mol-1) | Energy for Dihydro derivative 4(kcal mol-1) |
|---|---|---|
| Stretching | 1.2266 | 1.0963 |
| Bending | 18.8517 | 14.5074 |
| Torsion | 12.2412 | 12.4972 |
| Van der Waals | 5.7571 | 4.5124 |
| Dipole-dipole | 0.1630 | 0.1407 |
All the energy terms except the torsion energy of dihydro derivative 3 are higher than those of 4. The bending and van der Waals energies are specially higher in the dihydro derivative 3.
Stereochemitsry of nucleophilic additions to a pyridinium ring
Derivative of prolinol (5)
Models of pyridinium reactant 5 are energy-minimised using the MM2 force field. 10 different starting points are tried for optimization by moving one of the bonds randomly.
The MeMgI component cannot be added when minimising the models as the Mg atom has no atom type assigned.
The results for optimization are shown in the table below:
| Total energy (kcal mol-1) | Geometry | |
|---|---|---|
| 1 | 26.3220 | 
|
| 2 | 26.4193 | 
|
| 3 | 26.3394 | 
|
| 4 | 164.5849 | 
|
| 5 | 26.3411 | 
|
| 6 | 26.3362 | 
|
| 7 | 145.9397 | 
|
| 8 | 138.0110 | 
|
| 9 | 26.3719 | 
|
| 10 | 26.3143 | 
|
The models with 26.3 kcal mol-1 have the lowest energies and are most stable. It can be seen from the result that the seven-membered ring has very little flexibility and there are only two conformational minima possible.
They are shown as follows:
The carbonyl group and the aromatic pyridine ring are coplanar.
The carbonyl group is above the plane of the aromatic pyridine ring.
The pyridinium reactant 5, which is an N-methyl and N-benzyl derivatives of pyridoxazepinone, undergoes highly stereoselective addition with MeMgI, which is a Grignard reagent, due to reagent chelation.

When MeMgI is added to the pyridinium reactant 5, the Mg will chelate to the amide oxygen atom and this will lead to conjugate delivery of the Me group to the 4-position of the aromatic pyridine ring, forming a Mg enolate. As there is no possible conformation in which the carbonyl group is below the plane of the aromatic pyridine ring, it is only possible to have a product with an absolute stereochemistry of the Me group at the 4-position of the pyridine ring being anti to the H atom at the chiral centre of the L-prolinol auxiliary.
Key Literature
- A. G. Shultz, L. Flood and J. P. Springer, J. Org. Chemistry, 1986, 51, 838. DOI:10.1021/jo00356a016
Quinolinium Salt (7)
Models of the quinolinium salt 7 are energy-minimised using the same method as described for reactant 5.
The results for optimization are shown in the table below:
| Total energy (kcal mol-1) | Geometry | |
|---|---|---|
| 1 | 15.4884 | 
|
| 2 | 15.7895 | 
|
| 3 | 15.5363 | 
|
| 4 | 74.6199 | 
|
| 5 | 30.9663 | 
|
| 6 | 117.6712 | 
|
| 7 | 291.8098 | 
|
| 8 | 144.3389 | 
|
| 9 | 32.2949 | 
|
| 10 | 15.4885 | 
|
Model 1 has the lowest energy (15.4884 kcal mol-1) among all the models with different starting points and is most stable. The carbonyl group is behind the plane of the aromatic ring with N.
The simple model of the quinolinium salt 7 given in the question can be improved by indicating that the C=O bond is pointing into the plane (see the molecule below).

When aniline is added to the quinolinium salt 7, the pyridinium ring is derivatised, forming a corresponding 1,4-adduct 8. The NHPh group is then transferred to an electrophile, forming the desired product, PhHN-E, with the recovered quinolinium salt 7.

The quinolinium salt 7 is axially chiral with the chiral axis, C(3)-C=O, being the main configurational element. This is assured by the presence of the chiral inducer on the lactam moiety as the additional configurational element. The C(3)-C=O chiral axis is responsible for the high diastereoselectivity in the 1,4-reduction of the quinolinium salt (the “anchoring step” for the NPhH group).
Key Literature
- Leleu, Stephane; Papamicael, Cyril; Marsais, Francis; Dupas, Georges; Levacher, Vincent. Tetrahedron: Asymmetry, 2004, 15, 3919-3928 DOI:10.1016/j.tetasy.2004.11.004
Stereochemistry and Reactivity of an Intermediate in the Synthesis of Taxol
The isomers 10 and 11 are energy-minimised using the MM2 force field and the results are shown as follows:

An isomer with the carbonyl group pointing up and a twist-boat conformation on the fused cyclohexane.

An isomer with the carbonyl group pointing down and a chair conformation on the fused cyclohexane.
Isomers 10 and 11 are atropisomers as a hindered rotation about the single C-C=O bond will generate a high enough steric strain barrier and allow for the isolation of the conformers.
As isomer 11 is 10 kcal mol-1 lower in total energy than isomer 10, it is more stable. Thus, isomer 11, with the carbonyl group pointing down, is determined to be the key intermediate in the total synthesis of Taxol. Isomer 10 is high in energy because the bridged E-cyclononeone is so sufficiently rigidified that the adjoining fused cyclohexane (C ring) is locked, forming a twist-boat conformation.
According to literature, most of the oxy-Cope rearrangements are viewed as irreversible processes. However, the oxy-Cope rearrangement of the compound that is being tickled establishes an equilibrium.

The starting material undergoes [3,3] sigmatropy to form one of the two enol conformer with a large C ring, which then tautomerises into the ketone conformer. The ketone conformer in this case is thermodynamically comparable to the enol conformer and the starting material. Therefore, this process is reversible.

Production of which isomer depends on the nature of the substituents X and Y on the cyclohexene. The compound in this case is unsubstituted and only isomer 11 is formed. There are cases where both of the isomers are formed in the same process.
During subsequent functionalisation of the alkene, it reacts abnormally slowly because the cycloalkene in the compound is a “hyperstable alkene”. It is known that the Strain Energy (Est) of a cycloalkene is generally higher than that of its corresponding cycloalkane. In another words, cycloalkenes generally have positive Olefin Strain Energies (OSE). However, the hyperstable alkenes have positive OSE. The increase in Est is due to the increase in vicinal and transannular H interactions. Although the OSE of these hyperstable alkenes increases, their change in Enthalpy of formation, ∆Hf(alkene) - ∆Hf(alkane), are positive but lower than usual. Therefore, they are reluctant to be hydrogenated even under drastic catalytic conditions.
Key Literature
- S. W. Elmore and L. Paquette, Tetrahedron Letters, 1991, 319 DOI:10.1016/S0040-4039(00)92617-0
- J. G. Vinter and H. M. R. Hoffman, J. Am. Chem. Soc., 1974, 96, 5466 DOI:10.1021/ja00824a025
- Dale L. Boger, Susumu Miyazaki, Seong Heon Kim, Jason H. Wu, Olivier Loiseleur, and Steven L. Castle, J. Am. Chem. Soc., 1999, 121, 3226 DOI:10.1021/ja990189i
- Pelayo Camps, Francesc Perez and Santiago Vdzquez, Tetrahedron 1997, 53, 28, 9727-9734 DOI:10.1016/S0040-4020(97)00595-4
- Camps, P; Pujol, X; Vazquez, S, et al., Tetrahedron 2001, 57, 40, 8511-8520 DOI:10.1016/S0040-4020(01)00802-X
How one might induce room temperature hydrolysis of a peptide
Models of the two isomers 13 and 14 are energy-minimised using the MM2 force field and a chair-chair conformation as the template for the cis-decalin. The results are shown as follows:

An isomer with the ring N-substitutent orientated axial with respect to the decalin ring.
An isomer with the ring N-substitutent orientated equatorial with respect to the decalin ring.
Isomer 13 reacts faster than isomer 14 because its axially orientated OH group can approach the axially orientated carbonyl peptide bond close enough for hydrolysis to occur while this will be more difficult for isomer 14 as a lot more energy will be required for its distanced OH group and carbonyl peptide bond to react.
With careful conformational design, the compounds 13 and 14 undergo rapid room temperature esterification with half-lives of 21 and 840 minutes respectively while the half life for hydrolysis of a peptide bond at neutral pH and 25° is around 500 years.
The room temperature esterification of the compounds 13 and 14 is an intramolecular reaction, which proceeds via a cyclisation step invariably and is sometimes accompanied by a relief of strain in the molecule. Generally, intramolecular systems with a relief of steric strain react exceptionally fast. This explains for the rapid reaction rates of the two compounds as both of them are unstrained intramolecular systems with simple juxtapositions of the amide bonds and the catalytic functionality.
Key Literature
- M. Fernandes, F. Fache, M. Rosen, P.-L. Nguyen, and D. E. Hansen, J. Org. Chem., 2008, 73, 6413–6416 DOI:10.1021/jo800706y
- Dr. M. I. Page, Angew. Chem. lnr. Ed. Engl., 1977, 16, 7, 449-459
Modelling Using Semi-empirical Molecular Orbital Theory
Regioselective Addition of Dichlorocarbene
Part 1
The energy and geometry of compound 12 can be determined using the MM2 method. However, this is a purely mechanical treatment and is not useful at all when dealing with the electronic properties such as molecular strain, steric effects and stereoselectivity of a molecule. Therefore, it is explicit to taken into account the electrons in a molecule as they influence bonds and derived spectroscopic properties.
The result obtained using the MM2 method is shown as follows:

The MM2 method evaluate a molecule quickly but only provide quantitative results, e.g. the total energy, the stretching, bending, torsion and Van der Waals energy-terms of the molecule.
In order to look at the orbital control of reactivity in the reaction of compound 12 with electrophilic reagents such as dichlorocarbene or peracid, the geometry, the energy of the orbitals and their graphical forms have to be produced in a more qualitative manner. However, they cannot be obtained using the MM2 method.
When looking at the electronic properties of a molecule, the HF/STO-3G self-consistent-field MO method can be used to provide an approximate representation of the valence-electron molecular wavefunction. This transforms the purely mechanical treatment into a quantum mechanical treatment which includes the wave-description of the electrons.
The results obtained using the HF/STO-3G self-consistent-field MO method are shown as follows:
The calculation finishes at an energy of -548182.95 kcal mol-1 (-873.585857 Hartrees).
-
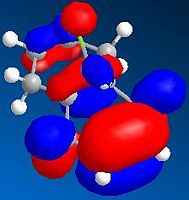 HOMO-1 (N=47, -8.449eV)
HOMO-1 (N=47, -8.449eV) -
 HOMO (N=48, -8.144eV)
HOMO (N=48, -8.144eV) -
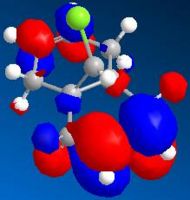 LUMO (N=49, 8.572eV)
LUMO (N=49, 8.572eV) -
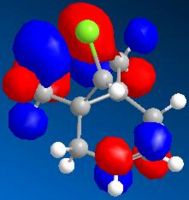 LUMO+1 (N=50, 8.980eV)
LUMO+1 (N=50, 8.980eV) -
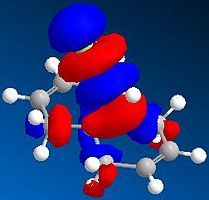 LUMO+2 (N=51, 9.736eV)
LUMO+2 (N=51, 9.736eV)





By comparing the models of compound 12, it can be seen that the electron density of the HOMO is most concentrated among the five orbitals shown. Thus, the HOMO is most nucleophilic and most reactive towards the electrophilic attack. As the electron clouds of the HOMO are concentrated on the cyclohexene ring syn to the C-Cl bond, the electrophilic reagents such as dichlorocarbene or peracid will react regiospecifically with compound 12 on the double bond syn to the C-Cl bond and on the face of the molecule opposite to the cyclopropyl ring, which can be reached more easily.
A more accurate Density-functional B3LYP/6-31G(d) approach can also be used to calculate the energy and the geometry of compound 12.
Part 2
Models energy-minimised using the MM2 force field:

Models of the hydrogenated version of Compound 12 with the anti double bond replaced by a C-C single bond are energy-minimised using the HF/STO-3G self-consistent-field MO method. The results as shown as follows:
-
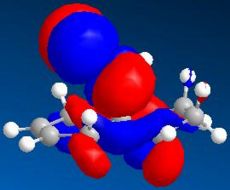 HOMO-1 (N=48, -9.000eV)
HOMO-1 (N=48, -9.000eV) -
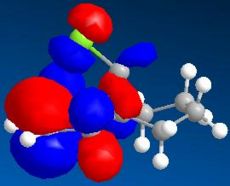 HOMO (N=49, -8.103eV)
HOMO (N=49, -8.103eV) -
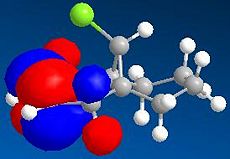 LUMO (N=50, 8.994eV)
LUMO (N=50, 8.994eV) -
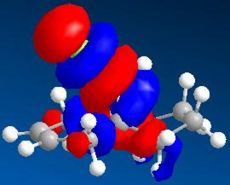 LUMO+1 (N=51, 9.735eV)
LUMO+1 (N=51, 9.735eV) -
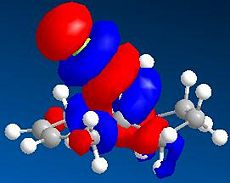 LUMO+2 (N=52, 12.254eV)
LUMO+2 (N=52, 12.254eV)





Key Literature
- B. Halton, R. Boese and H. S. Rzepa., J. Chem. Soc., Perkin Trans 2, 1992, 447 DOI:10.1039/P29920000447
Structure based Mini project using DFT-based Molecular orbital methods
Objectives and Suggested Structural Explorations
Regio- and stereoselective conversion of alkenes to epoxides


NMR spectroscopy can be used to ensure that the “correct” alkene had been epoxidised.