
Rep:Mod:eg1511
Experiment 1C involves the use of computer modeling to predict and explain the geometry and regioselectivity of certain molecules and 1H and 13C NMR. The asymmertric epoxidation involving the Shi and Jacobsen catalyst and the alkenes stilbene and 1,2-dihydronapthalene will be investigated, including prediction of their configuration, the oxide 1H and 13C NMR and interactions in their active sites.
Cyclopentadiene dimers
Cyclopentadiene dimers
This calculation involves investigating why the endo cyclopentadiene molecule is the major product of the Diels-Alder dimerisation of cyclopentadiene. In this reaction clyclopentadiene acts as both the diene and dienophile. This reaction is slightly unusual, it is a bispericyclic reaction and has characteristics of both π2s + π4s and a π4s + π2s cycloadditions and also a [3,3] sigmatropic shift [1].
The geometry of each cyclopentadiene dimer was optimised using Avagadro and the MMFF94s mechanics force field. The following energies were calculated.
| 1 | 2 | |
|---|---|---|
| Total bond stretching energy kcal/mol | 4.54399 | 3.46791 |
| Total angle bending energy kcal/mol | 30.77260 | 33.18934 |
| Total torsional energy kcal/mol | -2.73095 | -2.94947 |
| Total Van der Waals energy kcal/mol | 12.80161 | 12.35873 |
| Total electrostatic energy kcal/mol | 13.01367 | 14.18451 |
| Total energy kcal/mol | 55.37342 | 58.19067 |
Molecule 1 |
Molecule 1 |
The table above shows the total energies for Molecule 1 (top Jmol image) and Molecule 2 (bottom Jmol image), the exo and endo cylopentadiene dimers respectively. It shows that the exo dimer is lower in energy and is therefore the thermodynamic product, and the endo is the kinetic product. In the dimerisation of clyclopentadiene, the endo dimer is the exclusive product indicating that this reaction is under kinetic control.

The energies of the transition states can also be inspected.
| Dimer | TS energy Hartrees [2] | TS energy kcal/mol |
|---|---|---|
| 1 | -385.5131284 | -241913.1920929 |
| 2 | -385.5169885 | -241915.6143428 |
In the literature, basis set 6-31G*// 6-31G* was used. The energy of the transition states are not as expected. The transition state for molecule 1 (exo dimer) is higher in energy than for molecule 2 (endo dimer). This does not follow the trend for the energy of the products, however, the energy difference between the molecules is very small so this does not have much effect, and the kinetic factors dominate the reaction.

The reason why the endo product is the only product can be explained by looking at the HOMO-LUMO frontier orbitals and the secondary (non-bonding) orbital interactions (SOIs). In the endo transition state there is a bonding interaction between the carbonyl groups of the dienophile and the π bond forming at the back of the diene, this is the primary orbital interaction. There is also a secondary interaction between orbitals at the back of the diene, that are not directly involved in bonding but have a stabilising interaction. The exo transition state has the primary orbital interaction but lacks the stabilisin secondary orbital overlap. Due to this the endo product is formed more quickly and is therefore the only product formed. [4].
Hydrogenation of the cyclopentadiene dimers
Molecule 2 can be partially hydrogenated to form Molecules 3 and 4. The energies of the molecules can be determined by optimising the geometries using the MMFF94s mechanics force field.
| 3 | 4 | |
|---|---|---|
| Total bond stretching energy kcal/mol | 3.31162 | 2.82313 |
| Total angle bending energy kcal/mol | 31.94312 | 24.68533 |
| Total torsional energy kcal/mol | -1.47870 | -0.37841 |
| Total Van der Waals energy kcal/mol | 12.80161 | 10.63733 |
| Total electrostatic energy kcal/mol | 5.11948 | 5.14702 |
| Total energy kcal/mol | 50.44567 | 41.25749 |
These calculations show that Molecule 4 is more stable than Molecule 3 by 9.18818 kcal/mol, and hence 4 is the thermodynamic product and 3 is the kinetic product. 4 is more stable as it doesn't have a double bond close to the bridging carbon, which reduces the total angle strain in the molecule. 3 has a more negative torsional energy, as the more strained sp2 carbon is only 1 carbon away from the strained bridging group, compared to 4, where the sp2 carbon in the alkene is 2 atoms away. 3 also has higher bond stretching and angle bending energies as the double bond is within the cyclohexane ring, increasing its ring strain. Whereas the double bond in 4 is not in the cyclohexane ring, meaning that all the contributions except the electrostatic energies do not deviate from 'normality' as much.
References for this section
- ↑ Henry Rzepa http://www.ch.ic.ac.uk/local/organic/pericyclic/p1_categories.html
- ↑ 2.0 2.1 W. L. Jorgensen, D. Lim, and J. F. Blake, "Ab Initio Study of Diels-Alder Reaction of Cyclopentadiene with Ethylene, Isopropene, Cyclopentadiene, Acrylonitrile, and Methyl Vinyl Ketone", J. Am. Chem. Soc.,1993, 115, 2936-2942.DOI:10.1021/ja00060a048 10.1021/ja00060a048 Cite error: Invalid
<ref>tag; name "ja00060a048" defined multiple times with different content - ↑ http://openwetware.org/wiki/Todd:Chem3x11_ToddL13, date accessed: 30/01/13
- ↑ J. Clayden, N. Greeves, S. Warren and P. Wothers, Organic Chemistry, Oxford University Press, New York, 9th edn, 2011, p916.
Atropisomerim
The following molecules 9 and 10 are intermediates in the synthesis of Taxol. They are atropisomers of each other as they are isomers of each other due to restricted rotation about the carbonyl bond. The carbonyl can either can either point up (molecule 9) or down (molecule 10). The MMFF94s mechanics force field in Avagadro was used to optimise the geometry of both molecules and find the energies and hence the more stable isomer.
| 9 | 10 | |||||||
| Twist boat 1 | Twist boat 2 | Chair 1 | Chair 2 | Twist boat 1 | Twist boat 2 | Chair 1 | Chair 2 | |
| Total bond stretching energy kcal/mol | 7.93774 | 7.58208 | 7.68915 | 7.55380 | 7.75019 | 7.86433 | 8.83167 | 7.58944 |
| Total angle bending energy kcal/mol | 29.55659 | 21.90814 | 28.29266 | 21.58522 | 19.04323 | 21.04456 | 22.3460 | 18.83269 |
| Total torsional energy kcal/mol | 2.74862 | 2.12483 | 0.16464 | -0.58900 | 3.72867 | 4.45325 | 5.33874 | 0.18620 |
| Total Van der Waals energy kcal/mol | 34.67848 | 34.83406 | 33.18540 | 33.50193 | 35.01114 | 34.68705 | 36.42645 | 33.29534 |
| Total electrostatic energy kcal/mol | 0.31576 | 0.29185 | 0.29661 | 0.277738 | -0.06624 | -0.04724 | 0.38566 | -0.05727 |
| Total energy kcal/mol | 76.28448 | 67.78408 | 70.54097 | 63.29997 | 66.30589 | 68.89546 | 74.80417 | 60.55564 |

Molecules 9 and 10 were optimised using the MMFF94s mechanics force field. Molecule 9 has the carbonyl pointing upwards whereas in 10, the carbonyl group is pointing downwards. Each molecule also has different conformers due to the clyclohexane ring. Here the two chair and two boat conformations have been modeled. In both molecules the chair conformation is the lowest, this is as expected and can be seen in the diagram on the right. This is an energy profile diagram which shows the relative energies of the different conformations of cyclohexane. There are more conformations of cyclohexane such as the half chair which have not been modeled as they are even higher in energy. The diagram also shows more clearly the structure of the chair and twist boat conformers and the differences in structure between each chair and twist boat. It is also easy to see that the twist boat structures are more twisted and therefore will have higher torsional and angle bending energies. When looking at the data in the table above we can see that this is generally true. The 'chair 2' conformation of 10 is more stable than the 'chair 2' conformation of 9 by 2.74357 kcal/mol, which indicates that 10 is the more stable conformer and hence the thermodynamic product.
Normally, alkenes react readily however, these molecules react much more slowly than expected. These types of bridged alkenes are known as hyperstable alkenes and are thermodynamically very stable. They react slowly as the alkene is attached to a bridging group. A bridged alkene is very stable to reaction as it is less strained than the saturated hydrocarbon (olefinic strain) due to the double bond and residual strain due to the carbon skeleton. This means that they have negative olefinic strain relative to the alkane molecule. [2]. The formation of a bridgehead alkene leads to changes in structure and reductions in energy. When looking at the difference in energy between a bridged alkene and the saturated version of the same molecule. There are significant changes in energy in the angle bending, torsional and Van der Waals component, with the angle bending energy contributing the largest amount. We expect the alkene to have less strain in these components compared to the alkane. A bridged alkene flattens the bridgehead carbon relative to the alkane which also relieves the angle strain. The double bond is sp2 hybridised compared to the sp3 center of an alkane and this makes the double bond less sterically crowded[2].
References for this section
- ↑ J. Clayden, N. Greeves and S. Warren, Organic Chemistry,Oxford University Press, New York, 2nd edn, 2001, p373.
- ↑ 2.0 2.1 J. Kim, "The Molecular Mechanics Evaluation of the Stability of Bridgehead Olefins Containing Medium Rings", J. Am. Chem. Soc., 1981, 103, 1891.DOI:10.1021/ja00398a003 Cite error: Invalid
<ref>tag; name "ja00398a003" defined multiple times with different content
Both molecules 17 and 18 were drawn in ChemBio3D and the geometries were minimised using the MMFF94s mechanics force field. The lowest energy conformation for both intermediates was calculated and it was found that 18 was lower in energy and hence the thermodynamic product.
| Molecule 17 | Molecule 18 | |
|---|---|---|
| Total bond stretching energy kcal/mol | 15.94750 | 14.39222 |
| Total angle bending energy kcal/mol | 31.40455 | 28.60034 |
| Total torsional energy kcal/mol | 13.58620 | 13.55739 |
| Total Van der Waals energy kcal/mol | 53.27730 | 50.48944 |
| Total electrostatic energy kcal/mol | -7.10570 | -6.28394 |
| Total energy kcal/mol | 108.90205 | 102.27846 |
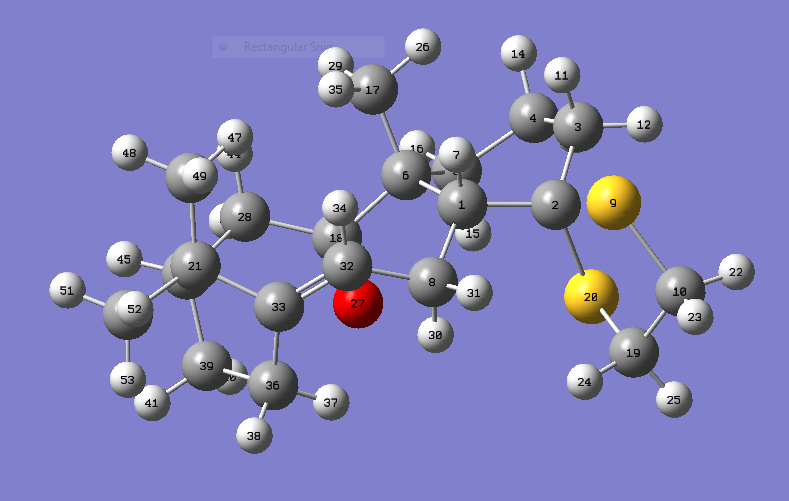{kind=link}
Molecule 18
18 is the more stable molecule, so the geometry at the density functional level (DFT) using Avodagro was calculated with this isomer. The geometry was optimised with the basis set B3LYP/6-31G(d,p), the solvent the calculation was run with was chloroform.From this calculation the 1H and 13C NMR were generated using Gaussian, the results have been published onto the D-Space [1]
1H NMR
An image of the molecule with labeled atoms for reference is provided here.
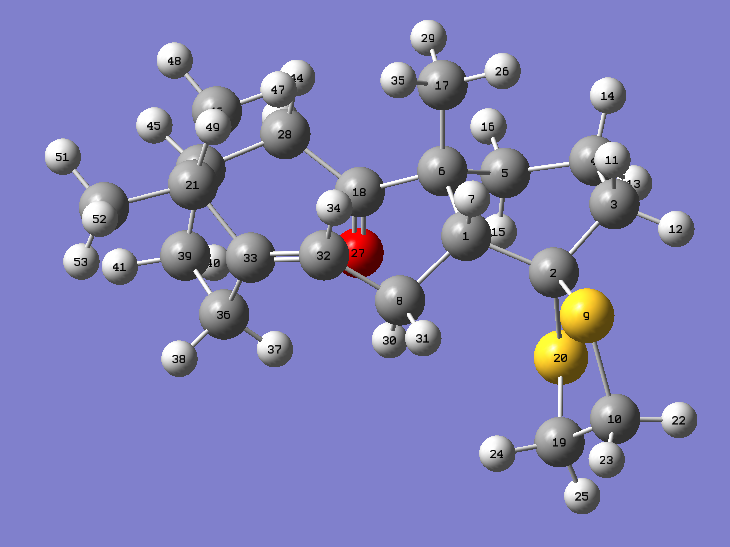{kind=link}
Below is the 1H NMR spectrum for molecule 18.

| Atom | Calculated chemical shift ppm | Integration | Literature shift ppm [2] | Multiplicity and integration |
|---|---|---|---|---|
| 51, 52 | 0.96 | 2H | 1.03 | (s, 3H) |
| 16, 29, 48 | 1.07 | 3H | 1.07 | (s, 3H) |
| 49 | 1.16 | 1H | 1.10 | (s, 3H) |
| 26 | 1.35 | 1H | ||
| 43 | 1.41 | 1H | ||
| 47, 53 | 1.50 | 2H | 1.20-1.50 | (m, 3H) |
| 14 | 1.6 | 1H | ||
| 13, 35 | 1.68 | 2H | 1.58 | (t, J = 5.4 Hz, 1H) |
| 7, 41 | 1.85 | 2H | 1.70-2.20 | (m, 6H) |
| 38, 45 | 1.96 | 2H | ||
| 37 | 2.09 | 1H | ||
| 11, 12, 15 | 2.27 | 3H | 2.35-2.70 | (m, 4H) |
| 30 | 2.40 | 1H | ||
| 40 | 2.46 | 1H | ||
| 31, 44 | 2.86 | 2H | 2.70-3.00 | (m, 6H) |
| 25, 33 | 3.12 | 2H | ||
| 22 | 3.22 | 1H | ||
| 24 | 3.32 | 1H | ||
| 34 | 5.28 | 1H | 5.21 | (m, 1H) |
Comparing the computed 1H NMR values with those found in the literature, the values follow the same overall trend,the integrations are largely correct and most shifts are within 0.2 ppm of the literature values. There are some differences however. The main one being that in the literature many of the hydrogen chemical shifts are close together e.g. 2.7-3 ppm, 6H and so have been measured as a multiplet whereas Gaussian computes each shift individually. The measurements found in the literature were measured at 300MHz in deuterated chloroform and the computated values were calculated at an unknown frequency in chloroform. Hydrogen 34 has the highest chemical shift,it is attatched to a carbon atom which is part of a double bond which is an area of high electron density and so deshields the hydrogen atom leading to increased chemical shift. The literature value have been aligned with the calculated shifts. However, they do not match perfectly, of the chemical shifts of atoms 11, 12 and 15, two of these atoms lie within the 1.70-2.20 ppm, 6H multiplet and the third atom is part of the 2.35-2.70 4H multiplet. Due to this factor, this slightly skews how the top half of the data can be aligned with the calculated results.
13C NMR
Below is the 13C NMR spectrum for molecule 18. The solvent used was chloroform and the basis set was B3LYP/6-31G(d,p).

| Atom | Chemical δ ppm | Literature ppm [2] |
|---|---|---|
| 4 | 21.05 | 19.83 |
| 46 | 23.39 | 25.26 |
| 50 | 25.74 | 22.21 |
| 36 | 28.66 | 25.35 |
| 17 | 28.85 | 25.56 |
| 8 | 31.04 | 30.00 |
| 5 | 35.31 | 30.84 |
| 28 | 37.58 | 35.47 |
| 3 | 42.04 | 36.78 |
| 10 | 44.22 | 40.82 |
| 19 | 48.20 | 43.28 |
| 21 | 50.11 | 45.53 |
| 42 | 55.60 | 50.94 |
| 6 | 56.11 | 51.30 |
| 1 | 67.88 | 60.53 |
| 2 | 89.09 | 74.61 |
| 32 | 118.83 | 120.90 |
| 33 | 148.67 | 148.72 |
| 18 | 211.83 | 211.49 |
The measurements found in the literature were measured at 75MHz in deuterated chloroform and the computated values were calculated at an unknown frequency in chloroform. The calculated 13C shits follow the same general trends as those found in the literature. The shift for atom 18 is the highest as the carbon atom is connected to the oxygen atom as part of the carbonyl group, the oxygen is electronegative and so deshields the carbon atom much more than all the other carbon atoms. The chemical shifts for atoms 18 and 33, however there are larger deviations in the 13C NMR compared to the 1H NMR.
References for this section
- ↑ Emily Giversen , "Gaussian Job Archive for C20H30O1S2", 2014.DOI:10042/27210
- ↑ 2.0 2.1 L. A. Paquette, N. A. Pegg, D. Toops, G. D. Maynard and R. D. Rogers, "[ 3.31 Sigmatropy within 1 -Vinyl-2-alkenyl-7,7-dimethyl-exo-norbornan-2-01~. The First Atropselective Oxyanionic Cope Rearrangement", J. Am. Chem. Soc., 1990, 112, 211-283.DOI:10.1021/ja00157a043
Crystal structures of the catalysts
Shi catalyst
The conquest program was used to draw pre-cursors 21 and 23, and this was used to search the Cambridge crystal database. The Mercury program was used to measure the C-O bond lengths for the anomeric centres (O-C-O) bonds.

| Bond | Bond length Ångstoms |
|---|---|
| C7-O2 | 1.441 |
| C7-O1 | 1.413 |
| C2-O6 | 1.403 |
| C2-O2 | 1.403 |
| C10-O5 | 1.409 |
| C10-O4 | 1.439 |
The data shows that the bond length either side of the central carbon atom in an anomeric centre differs, they are not the same. This could be due to the anomeric effect. This occurs due to the nOsp3, the lone pair on an oxygen atom donating electron desinty with a σ*C-O acceptor orbital which stabilises the α anomer. A resonance structure forms in which the oxygen that donated a lone pair forms a double bond and the bond between the carbon atom and the other oxygen bond is broken. As well as the anomeric effect, it is also known as 'no-bond, double-bond resonance' due to these two separate resonance forms. So for the α anomer, we would expect the bond to have a degree of partial double bond character, we can see that the bonds C10-O5 and C7-O1 are shorter than C10-O4 and C7-O2, and so we would expect the oxygen atoms in these bonds to be in the α position (O5 and O1) which the other oxygen atoms would be in the β position. The largest anomeric effect can be seen in the C10-O5 bond which is by shortest bond. The C10-O4 bond could be longer that the C10-O5 bond as it is close to the oxygen atom on the carbonyl group. There could be an electrostatic repulsion and some level of steric crowding.
The bonds C2-O6 and C2-O2 were measured but on inspection C2 is not an anomeric centre. This can be seen when looking at the bond lengths as both these bonds were measured to be the same length. These two bonds are slightly shorter than all the anomeric bonds which would indicate that they have a larger degree of partial double bond character. This could be due to an inductive effect from the strongly electronegative oxygen atom on the carbonyl group nearby.
Jacobsen catalyst

Looking at the structure of the Jacobsen catalyst, most of the molecule is planar with the tertiary butyl groups sticking out above and below the plane of the molecule. T-butyl groups are very bulky and take up a lot of space, and so blocking many angles of attack near to them. This helps to increase the highly selective nature of the catalyst.
The distance between the hydrogen atoms on the t-butyl groups were measured (and can be seen). The diagram shows a variety of bond lengths measured between hydrogen atoms on the t-butyl groups. The groups have Van der Waals forces which will be attractive when >2.1Å and repulsive when <2.1Å. This is the sum of the radius of two Hydrogen atoms. We would expect the H-H bond length within a t-butyl group to be short and so have unfavourable repulsive interactions. When measured this was found to be the case, bond lengths were measured to be 1.567 and 1.569Å which are less than 2.1Å and so are very unfavourable and increase the energy of the catalyst. The diagram shows that the bond lengths between hydrogen atoms on different t-butyl are greater than 2.1Å and so form attractive interactions. The t-butyl groups are position on the ortho and para positions, the bond length between the ortho hydrogen and a hydrogen on the t-butyl group is 1.920 and 1.945Å which are shorter than the sum of two hydrogen Van der Waals radii and so these interactions are quite unfavourable.
Analysis of epoxides
Trans stilbene oxide
The 1H and 13C NMRs were predicted using the same process as for predicting the NMR spectra for the Taxol intermediates. Trans stilbene oxide was drawn and the geometry was optimised in Avogadro using the MMFF94 s mechanics force field. The NMRs were predicted using the B3LYP/6-31G(d,p) basis set in Gaussian [1].
An image of trans stilbene with the atoms labeled is provided here for reference.
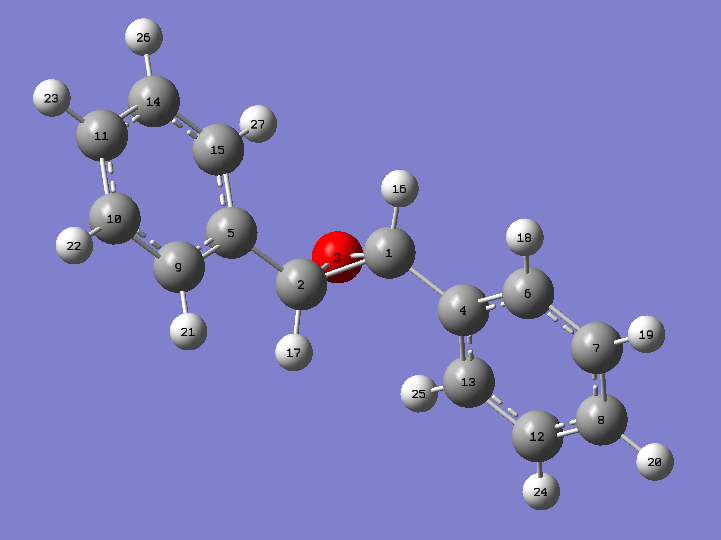{kind=link}
1H nmr
| Atom | Chemical shift ppm | Integration | Literature shift ppm [2] | Multiplicity and integration |
|---|---|---|---|---|
| 16, 17 | 3.54 | 2H | 3.84 | (s, 2H) |
| 18,21,19,22,23,20,25,27 | 7.48 | 8H | 7.28-7.37 | (m, 10H) |
| 24, 26 | 7.57 | 2H |
The literature only provides two chemical shifts for trans stilbene oxide which makes it difficult to assign the molecule and determine if the predicted 1H NMR is correct. The shifts around 7.3ppm are due to the hydrogens on the phenyl rings. When experimentally determined they all have a similar shift and the integration is a multiplet as there are long range coupling effects not making the integration and assignment simple. When calculated, Gaussian predicts these shifts for each Hydrogen atom induvidially. There are 10 hydrogens on the two phenyl rings which according to the literature are 7.28-7.37 ppm. The calculated values predicts 10 hydrogens in this shift area correctly. The shift at 3.54 is due to the two Hydrogens on the epoxide ring, they are considerably more shielded than the aromatic protons and therefore have a smaller chemical shift. The literature values were measured at 100MHz in deuterated chloroform. The calculated results were run in chloroform and at an unknown frequency.
13C nmr
| Atom | Chemical shift ppm | Literature shift ppm [2] |
|---|---|---|
| 1, 2 | 66.4 | 63.3 |
| 13, 15 | 118.3 | |
| 6, 9 | 123.1 | |
| 8, 11 | 123.2 | 126.0 |
| 7, 10 | 123.5 | 128.6 |
| 12, 14 | 124.2 | 129.3 |
| 4, 5 | 134.1 | 137.6 |
For 13C NMR the integration is not considered as the 13C abundancy is too low. Comparing the calculated results with data found in the literature, the results generally follow the same trend. The predicted values tend to differ to the literature by about 3 ppm. The shift at 66.43 ppm is about 3 ppm higher than the literature value, all the other checmical shifts are about 3 ppm lower than the literature. The literature has fewer shifts as these have been experimentally measured and the carbon atoms on the phenyl rings are not differentiated as much by either the NMR machine of the interpretation of the data. The shift at 66.53 is due to atoms 1 and 2 which are part of the epoxide ring, these have a smaller shift as they are not as shielded as the other atoms. The other chemical shifts are much higher, 123-134 ppm, these are due to the carbon atoms that lie in the two phenyl rings, they are more deshielded due to the delocalised π system in the phenyl rings. The literature data was measured at 100MHz in deuterated chloroform and the calculations were run at an unknown frequency in chloroform.
Optical rotation
The optical rotation for (1R, 1R) trans stilbene oxide were calculated as 363.18o(589 nm)and 1276.12o (365 nm) in chloroform, the results have been published to D-Space.[3]. The literature values found were 319.8o (589nm) in benzene [4]. No literature values were found for 365 nm. The calculated optical rotation is about 40o off the literature values which is not unreasonable as experimentally measured optical rotation measurements are known to vary and when searching the literature, a range of values were found. The literature values seem to vary with the temperature, the concentration and the solvent measured in. The literature values were measured in benzene and the calculated ORD was run in chloroform so this could be a reason why the two values differ.
1,2-dihydronapthalene oxide
The same process was carried out for 1,2-dihydronapthalene[5]. A labeled diagram of 1,2-dihydronapthalene is provided here for reference.
{kind=link}
Computated 1H NMR 1,2-dihydronapthalene oxide

| Atom | Chemical shift ppm | Integration | Literature shift ppm [6] | Multiplicity and integration |
|---|---|---|---|---|
| 18 | 1.56 | 1H | 1.77 | (1H, ddd, J=14.4, 14.4, 5.4 Hz) |
| 19 | 2.22 | 1H | 2.39-2.44 | (1H, m) |
| 16 | 2.27 | 1H | 2.55 | (1H, dd, J=14.4, 5.4 Hz) |
| 17 | 2.88 | 1H | 2.79 | (1H, ddd, J=14.4, 14.4, 6.4 Hz) |
| 20, 21 | 3.59 | 2H | 3.73-3.74 | (1H, m) |
| 3.85 | (1H, 4.5 Hz) | |||
| 14 | 7.20 | 1H | 7.09 | (1H, d, J=7.4 Hz) |
| 12, 13 | 7.39 | 2H | 7.20 | (1H, J=7.4, 7.4 Hz) |
| 7.26 | (1H, dd, J = 7.4, 7.4 Hz) | |||
| 15 | 7.49 | 1H | 7.39 | (1H, d, J=7.4 Hz) |
The table above shows that the predicted NMR follows the same trend as the literature. The chemical shifts are within about 0.3 ppm of the literature values. The chemical shifts for atoms 12-15 are above 7 ppm, they are much higher than the other shifts as tehy are part of the aromatic ring which deshields them. The next highest chemical shifts are for atoms 20 and 21, which are connected to the carbons atoms in the expxide ring. The elctronegative oxygen has an inductive electron withdrawing effect and deshields these atoms leading to a higher chemical shift. The electron withdrawing effect of oxygen is not as strong as the deshielding strength of an aromatic ring. The lower shifts are all normal for a hydrocarbon environment. The literature was measured at 500MHz in deuterated chlorform and the calculations were measured in chloroform at an unknown frequency.
Computated 13C NMR 1,2-dihydronapthalene oxide

| Atom | Chemical shift ppm | Literature shift ppm [6] |
|---|---|---|
| 8 | 24.9 | 21.8 |
| 7 | 28.0 | 24.4 |
| 10 | 54.8 | 52.7 |
| 9 | 56.4 | 55.1 |
| 1 | 121.4 | 126.1 |
| 2 | 123.4 | 128.4 |
| 3 | 123.8 | 128.4 |
| 6 | 125.6 | 129.5 |
| 5 | 130.4 | 132.5 |
| 4 | 133.8 | 136.7 |
The chemical shifts for carbons 1-6 are high 121.4-133.8 ppm. They are within the aromatic ring which deshields these carbons hence producing a large chemical shift. The lowest chemical shifts are for atoms 7 and 8 which are not part of an aromatic ring and not attached to the oxygen atom. Atoms 9 and 10 have chemical shifts of about 55 ppm in between the other two groups, they are connected to the oxygen atom which is electronegative and so deshields these carbon atoms. The electron withdrawing effect of the oxygen is not as strong as the deshielding effect of an aromatic ring, this is reflected in the lower chemical shifts. The predicted values follow the trend found in the literature, there are deviations of up to 4 ppm. The literature was measured at 125MHz in deuterated chlorform and the calculations were measured in chloroform at an unknown frequency.
Optical rotation
The optical rotation for (1R, 2S) 1,2-dihydronapthalene oxide was calculated in Gaussian and this gave an optical rotation of 52.77o (589nm) and 250.36o (365nm) in chloroform [7]. The literature value found is -39° at 589nm in chloroform for (1S, 2R) 1,2-dihydropnapthalene oxide [8]. This shows that the calculated molecule is the opposite enantiomer than the literature molecule. The 1H and 13C NMR results will be the same, and the ORDs should be the same expect with the opposite sign. Ignoring the sign of the ORD, the calculated value is not that far off the literature value. Optical rotation measurements are know to be quite unreliable and when searching the literature, quite a range of values were found. The literature values seem to vary with the temperature, the concentration and the solvent measured in. No literature value for 365 nm.
β-methyl styrene transition state
Shi catalyst
The energies of the transition states for β-methyl styrene with the Shi catalyst were used to find the free energy difference between the diasteroisomeric transition states, K, which is the ratio of the concentrations of the isomers and finally the enantiomeric excess of the major epoxide.
The transition states for β-methyl styrene with the Shi catalyst were given as this would take a long time to calculate. The sum of the electronic and thermal free energies for the 4 isomers for each R,R and S,S enantiomer.
| Transition state | Total energy Ha | Total energy KJ/mol | |
|---|---|---|---|
| RR | 1
|
-1343.022970
|
-3526107.0763
|
2
|
-1343.019233
|
-3526097.2648
| |
3
|
-1343.029272
|
-3526123.6222
| |
4
|
-1343.032443
|
-3526131.9477
| |
| SS | 1
|
-1343.017942
|
-3526093.8753
|
2
|
-1343.015603
|
-3526087.7343
| |
3
|
-1343.023766
|
-3526109.1662
| |
4
|
-1343.024742
|
-3526111.7287
|
These energies are the total transition state energy for each system, they have already been corrected for entropy, zero-point thermal energies and a solvation correction for water as a solvent.
From the above table of energies we can see that the lowest energy transition states are isomers RR 2 and SS 2. The difference in free energies between these states is ΔG= 0.007701 Ha or 20.218977 KJ/mol. From these values the equation:
This equation can then be rearranged to find k, where k is the ratio of the concentration of these two isomers. From this lnK was found to be -8.156697276 and K=0.000287.
The enantiomeric excess can be found using the equation:
The enantiomeric excess for β-methyl styrene is 0.999426548=99.94%. This shows that we would expect the R, R isomer to be the major product. This is slightly higher than the ee value found in the literature ee=95.7% [9], this shows how selective the Shi catalyst is for the asymmetric epoxidation of alkenes.
Jacobsen catalyst
The same process was carried out for the transition states of β-methyl styrene with the Jacobsen catalyst. The following energies were obtained
| Transition state | Total energy Ha | Total energy KJ/mol | |
|---|---|---|---|
| SR | 1
|
-3383.25956
|
-8882748.649
|
2
|
-3383.25344
|
-8882732.589
| |
| RS | 1
|
-3383.25106
|
-8882726.335
|
2
|
-3383.25027
|
-8882724.261
|
The lowest transition states are SR 1 and RS 1, the free energy difference is ΔG=0.008499 Ha or ΔG=22.314126 KJ/mol. Using the same equations as above, this gives lnK=-9.001917889 and K=0.000123. The enantiomeric excess was calculated ee=0.999754=99.98% in favour of the SR epoxide. The literature ee is 92%.[10] which is about 8% lower than the predicted value.
| Transition state | Total energy Ha | Total energy KJ/mol | |
|---|---|---|---|
| SS | 1
|
-3383.262481
|
-8882756.320518
|
2
|
-3383.257847
|
-8882744.153950
| |
| RR | 1
|
-3383.253816
|
-8882733.570559
|
2
|
-3383.254344
|
-8882734.956823
|
The lowest transition states are SS 1 and RR 2, the free energy difference is ΔG=0.008137 Ha or ΔG=21.363695 KJ/mol. lnK=-8.618497009 and so K=0.000181. This gives and enantiomeric excess ee=0.999639 =99.96% in favour of the SS epoxide. No literature was found for the enantiomeric excess of β-methyl styrene reacted with the Jacobsen catalyst.
Stilbene transition state
Shi catalyst
| Transition state | Total energy Ha | Total energy KJ/mol | |
|---|---|---|---|
| RR | 1
|
-1534.687808
|
-4029323.147
|
2
|
-1534.687252
|
-4029321.687
| |
3
|
-1534.700037
|
-4029355.254
| |
4
|
-1534.699901
|
-4029354.897
| |
| SS | 1
|
-1534.683440
|
-4029311.679
|
2
|
-1534.685089
|
-4029316.008
| |
3
|
-1534.693818
|
-4029338.926
| |
4
|
-1534.691858
|
-4029333.780
|
This table shows that the transitions states for 'RR 3' and 'SS 3' are the lowest in energy. The free energy difference is ΔG=0.006219 Ha or ΔG= KJ/mol, lnK=-6.587001851 and K= 0.001378. This gives and enantiomeric excess for the SS enantiomer ee=0.001378= 0.137817%. This is clearly not the enantiomer in excess. The enantiomer expected to form in excess is the RR enantiomer, ee = 100-0.001378 = 0.998622 = 99.86218% The literature ee for trans stilbene oxide is >95% trans stilbene RR [11] and so the calculated value correctly matches with the literature, although the literature does not give an exact value.
Jacobsen catalyst
| Total energy Ha | Total energy KJ/mol | |
|---|---|---|
| SS | -3574.921174
|
-9385956.257321
|
| RR | -3574.923087
|
-9385961.279903
|
From the total energies of these transition states, the free energy difference is ΔG=0.001913 Ha or ΔG=5.022582KJ/mol, lnK=-2.026199492, K=0.131836. From this ee=0.131836= 13.18356% This is the enantiomeric excess for the RR enantiomer, as above this is not the enantiomer is excess. The SS enantiomer is expected to form in excess ee=1-0.131836 = 0.868164 = 86.81644%. The literature found gives an ee=90% for SS cis trans stilbene[12]. This is closely matched to the literature value, it is about 3% lower than that in the literature.
Except for the above example, all the ee values calculated were in excess of 99%, which is exceptionally high, they are all higher than the literature values found. This could be because they are computated values and the literature values are experimentally determined. When doing experiment 1S, it is expected that the ee values will be considerably lower than both the literature values and the calculated values.
NCI
This part involves looking at the non-covalent interactions (NCIs) in the active site of the reaction transition state. Here the transition state for the reaction between Stilbene RR and the Shi catalyst. The first transition state was investigated, the NCI for this transition state was generated using Gaussview. Non-covalent interactions are not bonds within a molecule but intramolecular interactions. They can be defined as 'any relatively weak chemical bond that does not involve an intimate sharing of electrons. Multiple non-covalent bonds often stabilize the conformation of macromolecules and mediate highly specific interactions between molecules'[13]. In the diagram below the coloured in areas show the NCIs, where blue is very attractive (e.g. hydrogen bonding interactions, positive dipole-dipole interactions, favourable Van der Waals interactions, ionic interactions or π interactions), green is slightly attractive, yellow is slightly repulsive and red is very repulsive.
Orbital |
The multicoloured ring in the centre of the molecule is the bond forming in the transition state. It is comprised of both covalent and non-covalent interactions, it can also be thought of as half covalent. We can see that there are very few red areas, corresponding to very few repulsive interactions. The few red repulsive interaction that exist lie in the center of the two clyopentane rings. The two phenyl rings in trans stilbene both have a small amount of favourable yellow interactions. These could be due to the π cloud in the aromatic rings. Most of the interactions between the Shi catalyst and trans stilbene are blue and green indication this transition state has many favourable interactions and is hence a favourable reaction. The oxygen atoms are electronegative and so they will induce dipoles in the trans stilbene leading to induced dipole-induced dipole interactions. There could also be partial hydrogen bonding interactions between the oxygen atoms and the hydrogen atoms on the alkene.
QTAIM

It is not possible to obtain coordinates for the atoms but it is possible to analyse the diagram qualitatively.
The bond critical point (BCP) is the point in the elctron density curvature where the shared elctron density reaches a minimum[14]. These are represented in the QTAIM image yellow dots in between the pink atoms. The dotted lines are non covalent BCPs and generally lie in a similar position to the green and blue favourable interactions in the NCI, again showing that the interaction and following reaction with the catalyst is favourable. These BCPs appear where we would expect hydrogen bonding to occur between the oxygen atoms on the catalyst and the hydrogen atoms of the trans stilbene. The BCPs connected by solid lines represent what we call bonds and so are covalent in nature. It is interesting to note that the non-covalent BCPs lie mainly in the middle between two nuclei, whereas the covalent BCPs connecting hydrogen and carbon nuclei tend to lie closer to the hydrogen nucleus, showing that more electron density lies closer to the carbon nucleus than the hydrogen.
References for this section
- ↑ Emily Giversen , "Gaussian Job Archive for C 14H12O1", 2014.DOI:10042/27257
- ↑ 2.0 2.1 B. R. B Page, B. R. Buckley, D. Barros and J. Blacker, "Non-aqueous iminium salt mediated catalytic asymmetric epoxidation", Tetrahedron,2006, 62, 6607–6613.DOI:10.1016/j.tet.2005.10.084
- ↑ Emily Giversen , "Gaussian Job Archive for C14H12O1", 2014.DOI:10042/27253
- ↑ B. Wang, X. Wu, O. A. Wong. B. Nettles, M. Zhao, D. Chen and Y. Shi, "A Diacetate Ketone-Catalyzed Asymmetric Epoxidation of Olefins", J. Org. Chem.,2009, 74, 3986-3989 DOI:10.1021/jo900330
- ↑ Emily Giversen , "Gaussian Job Archive for C14H10O1", 2014.DOI:10042/27264
- ↑ 6.0 6.1 T. Niwa and M. Nakada, "A Non-Heme Iron(III) Complex with Porphyrin-like Properties That Catalyses Asymmetric Epoxidation", J. Am. Chem. Soc.,2012, 134, 13538-13541.DOI:10.1021/ja304219s
- ↑ Emily Giversen , "Gaussian Job Archive for C10H10O1", 2014.DOI:10042/27235
- ↑ H. Lui, J. Qiaoa, Y. Liua and Z. Wua, "Styrene monooxygenase from Pseudomonas sp. LQ26 catalyzes the asymmetric epoxidation of both conjugated and unconjugated alkenes", Journal of Molecular Catalysis B: Enzymatic,2010, 67, 236–241.DOI:10.1016/j.molcatb.2010.08.012
- ↑ Z. Wang, Y. Tu, M. Frohn, J. Zhang and Y. Shi, "An Efficient Catalytic Asymmetric Epoxidation Method", J. Am. Soc.,1997, 119, 11224-11235.DOI:10.1021/ja972272g
- ↑ E. N. Jacobsen, W. Zhang, A. R. Muci, J. R. Ecker and L. Deng, "Highly enantioselective epoxidation catalysts derived from 1,2-diaminocyclohexane", J. Am. Chem. Soc.,1991, 113, 7063–7064.DOI:10.1021/ja00018a068 10.1021/ja00018a068
- ↑ Y. Tu, Z. Wang and Y. Shi, "An Efficient Asymmetric Epoxidation Method for trans-Olefins Mediated by a Fructose-Derived Ketone", J. Am. Chem. Soc.,1996, 118, 9806-9807.DOI:10.1021/ja962345g
- ↑ S. Chang, J. M. Galvin and E. N. Jacobsen, "Effect of Chiral Quaternary Ammonium Salts on (salen)Mn-Catalyzed Epoxidation of cis-Olefins. A Highly Enantioselective, Catalytic Route to Trans-Epoxides", J. Am. Chem. Soc.,1994, 116, 6937-6938.DOI:10.1021/ja00094a059
- ↑ J. E. Darnell, H. Lodish. A. Berk. L. Zipursky, P. Matsudaira and D. Baltimore, Molecular Cell Biology, W.H.Freeman, New York, 4th edn, 2000, p212.
- ↑ B. Bankiewicz, Piotr Matczak and M> Palusiak, "Electron Density Characteristics in Bond Critical Point (QTAIM) versus Interaction Energy Components (SAPT): The Case of Charge-Assisted Hydrogen Bonding", J. Phys. Chem. A,2012, 116, 452–459.DOI:10.1021/jp210940b
Suggested alkenes for further study

A search was done on Reaxys to find epoxides with a molecular weight less that 200, and with an optical rotation smaller than -300oand larger than 300o. Pulgone has been chosen as an alkene for further study. It is available on Sigma-Aldrich, and is a naturally occuring essential oil found in plants such as peppermint[1] and it is also toxic to rats[2].
Pulegone has an optical rotatory power of 43.2o in ethanol, at λ=589nm at 25.0o [3]. When neat(no solvent), the optical rotation is 22o, λ=589 nm, 20oC[4]. From this we can see that the solvent has an effect on the optical rotation of the molecule. The difference between no solvent and ethanol is almost double. At λ=546.1nm, the optical rotation was measured to be 30o at 20oC in ethanol[5]. This shows that measuring the optical rotation at different wavelengths does not seem to change the optical rotation by very much which is quite unusual. This alkene could be studied to further detail to look how the optical rotation varies with wavelength and the solvent used.
References for this section
- ↑ D. R. Farley and V. Howland, "The natural variation of the pulegone content in various oils of peppermint, J. Sci. Food Agric., 2006, 31, 685-688.DOI:10.1002/jsfa.2740311104
- ↑ Z. Li, X. Xing, Z. Feng, C. Wu, X. Hou, Z. Xie and Q. Liao, "Absorption of volatile components of Kegan Liyan extract in rats by GC-MS", Zhongguo Yiyuan Yaoxue Zazhi, 2013, 33, 1041-1044.
- ↑ T. M. Feeley and M. K. Hargreaves, "Circular dichroism studies. The pulegone oxides", J. Chem. Soc,1970, C,1745-1750.DOI:10.1039/J39700001745
- ↑ D. A. Lightner and B. V. Grist, "Steric Inhibition of π-Electron Delocalisation in 1,3-Dienes ", Tetrahedron, 1981, 37, 685-688.DOI:10.1016/S0040-4020(01)97684-7
- ↑ F. Soccolini, G. Chelucci and C. Botteghi, "Synthesis of optically active 5,6,7,8-tetrahydroquinolines", J. Chem. Soc, 2009, C, 1001-1004.DOI:10.1002/jhet.5570210414