
Rep:Mod:ach2718
NH3 Molecule
Summary
| N-H bond length | 1.02Å |
| H-N-H Bond Angle | 106° |
| Calculation Method | RB3LYP |
| Basis set | 6-31G(d,p) |
| Final Energy E(RB3LYP) in au | -56.4439719 |
| RMS Gradient | 0.00000485 |
| Point Group | C3v |
Item Value Threshold Converged? Maximum Force 0.000004 0.000450 YES RMS Force 0.000004 0.000300 YES Maximum Displacement 0.000072 0.001800 YES RMS Displacement 0.000035 0.001200 YES
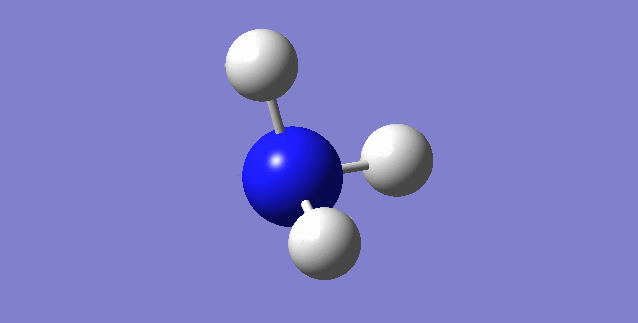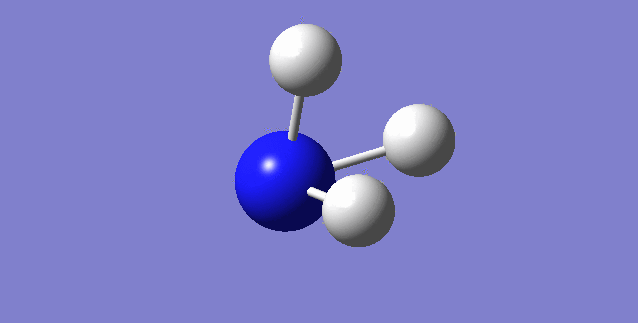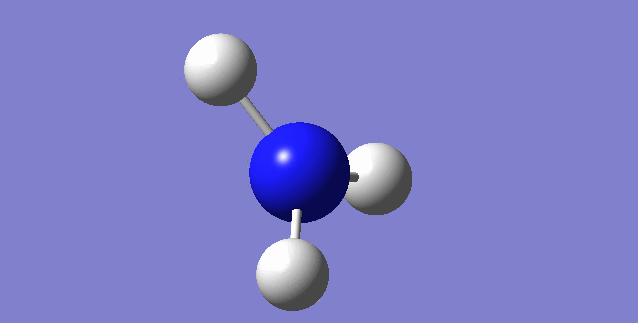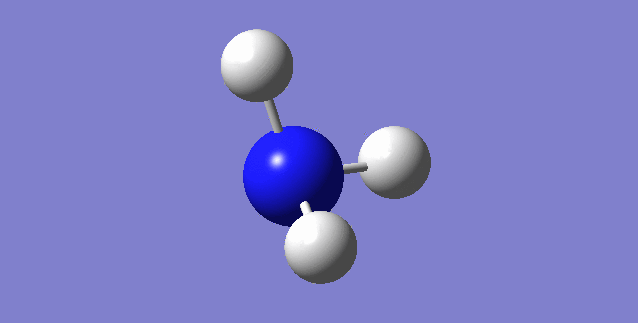
Jmol image and link to optimised structure file
NH3 dynamic image |
The optimisation file is liked to here
Vibration Information

| Wavenumber cm-1 | Symmetry | Intensity au | Image of Vibration | Video of Vibration |
|---|---|---|---|---|
| 1090 | A1 | 145 |  |
Click for the video here |
| 1694 | E | 14 |  |
Click for the video here |
| 1694 | E | 14 |  |
Click for the video here |
| 3461 | A1 | 1 |  |
Click for the video here |
| 3590 | E | 0 |  |
Click for the video here |
| 3590 | E | 0 |  |
Click for the video here |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
From the 3N-6 rule, you would expect 6 vibrational modes, which there are for NH3 molecule, being a non linear molecule. However, there are only four distinct wavenumber values and therefore both modes at 1694 and both vibrational modes at 3590 are degenerate. Since the two degenerate modes at 3590 have an IR intensity of 0 you would expect to see 3 bands in the IR spectrum. Bending vibrational modes are often lower in frequency than stretching, and NH3 follows this trend. The lowest three frequencies are bending and the highest three frequencies are stretches. From the image it is clear that vibrational mode four, at 3561 cm-1 with symmetry label A1, is the only symmetric stretch, the highly symmetric mode. An important bending mode ammonia undergoes is that known as the umbrella mode, which is mode the mode at 1090 cm-1 with the symmetry label A1.
Charge Analysis

The nitrogen in ammonia is shown in red and has a negative charge due to the lone pair of electrons not shown in the image. The negative charge on the nitrogen is enhanced by the fact it is electronegative, and therefore attracts the electrons in the covalent bond with hydrogen towards itself and therefore away from the hydrogen. This gives the hydrogen, shown in red, the partial positive charge we can see. The overall charge of the molecule is zero and therefore the positive and negative charges must cancel each other out.
N2 Molecule
Summary
| N-N Bond Length | 1.11 Å |
| N-N Bond Angle | 180° |
| Calculation Method | RB3LYP |
| Basis Set | 6-31G(d,p) |
| Final Energy E(RB3LYP) in au | -109.5241287 |
| RMS Gradient in au | 0.00000060 |
| Point Group | D∞h |
Item Value Threshold Converged? Maximum Force 0.000001 0.000450 YES RMS Force 0.000001 0.000300 YES Maximum Displacement 0.000000 0.001800 YES RMS Displacement 0.000000 0.001200 YES
Jmol image and link to optimised structure file
N2 Molecule image |
The optimisation file is liked to here
Vibration Information

| Wavenumber cm-1 | Symmetry | Intensity au | Image of Vibration |
|---|---|---|---|
| 2457 | SGG | 0 | 
|
Because this is a linear molecule, the number of vibrational modes expected is 3N-5, giving us the one seen above. The only mode is a stretching mode and there is no change in dipole, therefore no infrared radiation is absorbed by the molecule, so no signal is seen in infrared spectrometry.
Charge Analysis

As expected there is no charge on either molecule as N2 is a diatomic molecule and both nitrogens clearly have the same electronegativity, therefore neither pulls electrons within the bond. Due to this the bond is purely covalent, there is no ionic character at all.
Mono Metallic Transition Metal complex
Name: Hydrido-dinitrogen-tris(triphenylphosphine)-cobalt
Unique identifier:PPHCHN11
Dinitrogen bond length in complex: 1.11Å In the optimised calculated structure, the length of a diatomic nitrogen molecule is also 1.11Å. Therefore the bond lengths in the complex and the N2 are equal to 3 significant figures. It is expected that a more detailed analysis would show that the bond length in the complex is longer due to the metal weakening the bond. This is expected because in the coordinate bond electron density from the N-N bond is donated to the metal. However back donation could occur in certain complexes resulting in a stronger bond with shorter bond length in the complex.
The link for this molecule on CCDC is found here:- https://www.ccdc.cam.ac.uk/structures/Search?Ccdcid=PPHCHN11&DatabaseToSearch=Published
H2 molecule
Summary
| H-H Bond Length | 0.74Å |
| Bond Angle | 180° |
| Calculation Method | RB3LYP |
| Basis set | 6-31G(d,p) |
| Final Energy E(RB3LYP) in au | -1.1785394 au |
| RMS Gradient in au | 0.00000017 |
| Point Group | D∞h |
Item Value Threshold Converged? Maximum Force 0.000039 0.000450 YES RMS Force 0.000039 0.000300 YES Maximum Displacement 0.000052 0.001800 YES RMS Displacement 0.000073 0.001200 YES
Jmol image and link to optimised file
H2 optimisation |
The optimisation file is liked to here
Vibration Information

| Wavenumber cm-1 | Symmetry | Intensity au | Image of Vibration |
|---|---|---|---|
| 4466 | SGG | 0 |
H2 is also a linear molecule and therefore we expect one vibrational mode. It is again a symmetric vibration and therefore there is no change in dipole, and so no signal in the infrared spectrum.
Charge analysis

As H2 is another diatomic molecule with no electronegativity difference, it has no dipole and the bond is purely covalent.
Haber-Bosch Process
E(NH3)= -56.5577687 au
2*E(NH3)= -113.1155375 au
E(N2)= -109.5241287 au
E(H2)= -1.1785394 au
3*E(H2)= -3.5356181 au
ΔE=2*E(NH3)-[E(N2)+3*E(H2)]= -0.0557907 au
ΔE = -146.5kJmol-1
Ammonia is more stable than hydrogen and nitrogen gas which can be seen in the negative overall enthalpy change of -146.5 kJmol-1.
HCN
Summary
| C-H bond length | 1.068 Å |
| C-N Bond length | 1.16 Å |
| Calculation Method | RB3LYP |
| Basis set | 6-31G(d,p) |
| Final Energy E(RB3LYP) in au | -93.4245813 |
| RMS Gradient in au | 0.00017 |
| Point Group | C∞v |
Item Value Threshold Converged? Maximum Force 0.000370 0.000450 YES RMS Force 0.000255 0.000300 YES Maximum Displacement 0.000676 0.001800 YES RMS Displacement 0.000427 0.001200 YES
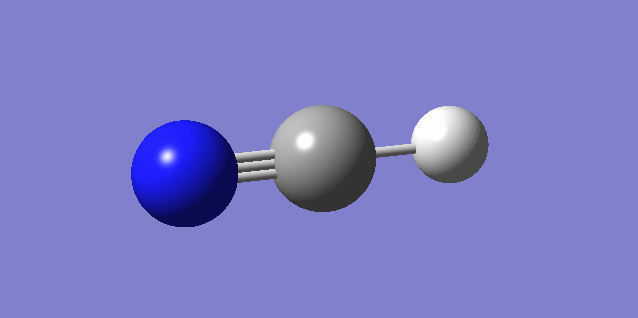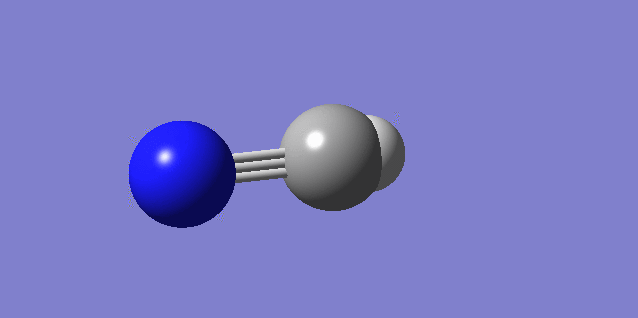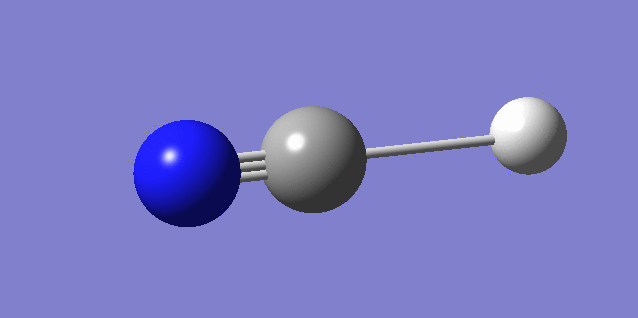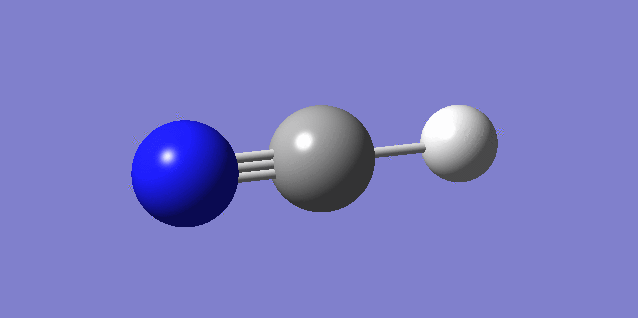
Jmol image and link to optimised file
HCN Molecule |
The optimisation file is liked to here
Vibration information

| Wavenumber cm-1 | Symmetry | Intensity au | Image of Vibration | Click for video of vibration |
|---|---|---|---|---|
| 766 | PI | 35 |  |
Click for video here |
| 766 | PI | 35 |  |
Click for video here |
| 2215 | SG | 2 |  |
Click for video here |
| 3480 | SG | 57 |  |
Click for video here |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Due to this being a linear molecule, we would expect 3N-5=4 vibrational modes. The two bending modes at 766 cm-1 with symmetry label PI are degenerate. As expected, the two bending vibrations are lower in wavenumber than the two stretching vibrational modes. The molecule does not have a centre of inversion so modes can be Raman active as well as infrared active. The two degenerate modes have a change of dipole moment which results in an infrared intensity of 35.
The stretching mode at 2215 cm-1 with symmetry label SG has only got an infrared intensity of 2. This is due to the nitrogen and hydrogen atom stretching symmetrically in opposite directions from the central carbon atom. The mode has an infrared intensity greater than zero because there is a triple bond between the nitrogen and the central carbon atom while there only is a single bond between the hydrogen atom and the central carbon atom. Due to the greater bond strength of the triple bond compared to the single bond, the hydrogen atom will be further away from the carbon atom than the nitrogen atom. Therefore the dipole moment will be slightly different to the equilibrium dipole moment.
The stretching mode at 3480 cm-1 with symmetry label SG has an infrared intensity of 57. The mode is highly infrared active because it is an asymmetric stretch as shown in the video. The dipole moment changes by a large extent due to the H-C bond distance being much longer in the stretch than the C-N bond distance in the stretch.
Charge Analysis

As expected the nitrogen has the negative charge due to the fact it is slightly more electronegative, and has still one lone pair on it, increasing its negative charge. The hydrogen has a slight positive charge only due to the small electronegativity difference between the carbon and hydrogen. This therefore means the molecule is polar.
Molecular Orbitals
| Molecular Orbital | Energy | Image | Main AO contributions | Bonding contribution |
|---|---|---|---|---|
| 1st Molecular Orbital | -14.36 | Nitrogen 1s | non bonding | |
| 5th Molecular Orbital | -0.38 |  |
Hydrogen 2s and 1s, Nitrogen 2s, 2pz, 3s and 3pz, Carbon 2pz and 2s | bonding |
| HOMO | -0.36 |  |
Nitrogen 2p and 3p, Carbon 2p and 3p | bonding |
| LUMO | -0.019 |  |
Nitrogen 2p and 3p, Carbon 2p and 3p | anti-bonding |
| 10th Molecular Orbital | 0.52 |  |
Hydrogen 2s and 1s, Nitrogen 2s, 2pz and 3s and 3pz, Carbon 2pz and 2s | anti-bonding |
The lowest energy MO has the main contribution from the Nitrogen 1s AO. There is almost no contribution from any other AOs which means that it is non bonding. In the picture it can be seen that there is no electron density shared between 2 atoms.
The 5th lowest MO has main contributions from the Hydrogen 2s and 1s, the Nitrogen 2s, 2pz, 3s and 3pz and the Carbon 2pz and 2s AOs where z is the direction along the bonds of the molecule. It is a σ bonding MO which can be seen from the diagram as there is electron density shared across all three atoms. The contribution of the nitrogen and hydrogen AOs are in phase with each other but out of phase with the carbon contribution.
The HOMO has main contributions from the Nitrogen 2p and 3p and the Carbon 2p and 3p AOs. The HOMO is degenerate with the 6th lowest MO. It is a π bonding MO which is seen clearly in the diagram by the large electron density above and below the central z axis through the bonds.
The LUMO has main contributions from the Nitrogen 2p and 3p and the Carbon 2p and 3p AOs. The LUMO is degenerate with the 9th lowest MO. It is the corresponding π anti-bonding MO to the HOMO. The diagram clearly shows the typical π antibonding shape with contributing AOs being out of phase above and below the central axis repelling each other.
The 10th lowest MO has main contributions from the Hydrogen 2s and 1s, the Nitrogen 2s, 2pz, 3s and 3pz and the Carbon 2pz and 2s AOs where z is the direction along the bonds of the molecule. It is the corresponding σ anti bonding MO to the 5th lowest energy MO. 3 phase boundaries are seen in the diagram making it much higher in energy than the 5th lowst MO.
This analysis was obtained using AO contributions to the MOs calculated by Gaussian. However, the AO contributions calculated do not perfectly represent the AOs present in the actual atoms.
Ethyne C2H2
Summary
| C-C bond length | 1.20 Å |
| C-H Bond length | 1.07 Å |
| Calculation Method | RB3LYP |
| Basis set | 6-31G(d,p) |
| Final Energy E(RB3LYP) in au | -77.3295039 |
| RMS Gradient in au | 0.00526 |
| Point Group | D∞h |
Item Value Threshold Converged? Maximum Force 0.000013 0.000450 YES RMS Force 0.000006 0.000300 YES Maximum Displacement 0.000023 0.001800 YES RMS Displacement 0.000013 0.001200 YES
Jmol image and link to optimised file
Ethene |
The optimisation file is liked to here
Use of hydrogen cyanide to form ethyne and diatomic nitrogen
2HCN → N2 + C2H2
E(N2)= -109.5241287 au
E(C2H2)= -77.3295039 au
E(HCN)= -93.4245813 au
2*E(HCN)= -186.8491626 au
ΔE=E(N2) + E(C2H2) - 2*E(HCN)= -0.00447 au
ΔE = -11.7kJmol-1
Marking
Note: All grades and comments are provisional and subject to change until your grades are officially returned via blackboard. Please do not contact anyone about anything to do with the marking of this lab until you have received your grade from blackboard.
Wiki structure and presentation 1/1
Is your wiki page clear and easy to follow, with consistent formatting?
YES
Do you effectively use tables, figures and subheadings to communicate your work?
YES
NH3 0.5/1
Have you completed the calculation and given a link to the file?
YES
Have you included summary and item tables in your wiki?
YES
Have you included a 3d jmol file or an image of the finished structure?
YES
Have you included the bond lengths and angles asked for?
YES
Have you included the “display vibrations” table?
YES
Have you added a table to your wiki listing the wavenumber and intensity of each vibration?
YES
Did you do the optional extra of adding images of the vibrations?
YES
Have you included answers to the questions about vibrations and charges in the lab script?
YES, most answers are correct. However there are only 2 visible peaks in the spectra of NH3, due to the low intensity of the other 2 peaks. (See infrared column in vibrations table.)
N2 and H2 0/0.5
Have you completed the calculations and included all relevant information? (summary, item table, structural information, jmol image, vibrations and charges)
YES, However you have given a bond angle of 180 for N2 and H2, there are no bond angles in diatomic molecules. Bond angles involve exactly 3 atoms.
Crystal structure comparison 0.5/0.5
Have you included a link to a structure from the CCDC that includes a coordinated N2 or H2 molecule?
YES
Have you compared your optimised bond distance to the crystal structure bond distance?
YES
Haber-Bosch reaction energy calculation 1/1
Have you correctly calculated the energies asked for? ΔE=2*E(NH3)-[E(N2)+3*E(H2)]
YES
Have you reported your answers to the correct number of decimal places?
YES
Do your energies have the correct +/- sign?
YES
Have you answered the question, Identify which is more stable the gaseous reactants or the ammonia product?
YES
Your choice of small molecule 5/5
Have you completed the calculation and included all relevant information?
YES, well done on the high level of detail in your vibrational analysis!
Have you added information about MOs and charges on atoms?
YES, again very detailed and well explained analysis well done!
Independence 1/1
If you have finished everything else and have spare time in the lab you could:
Check one of your results against the literature, or
Do an extra calculation on another small molecule, or
Do some deeper analysis on your results so far
You did an extra calculation on ethyne and you analysed the energetics of a relevant reaction independently, well done.