
Rep:Mod:ZQHMod1
Now it is possible to use computer to model many organic molecule structure and reactivity accurately. The modelling can not only explain the reactivity and the reaction mechanism of existing molecules, but also predict and develope the reaction and molecules. The properties of molecules can be expressed in terms of a summation of individual bond properties, for example, the bond stretching, bending, and torsion, van der Waal's repulsion and dipole dipole interactions. This method is known as Molecular Mechanics approach (MM). MM assumes the energy of a molecular system contains five individual terms as mentioned above. To be specific:
1. Diatomic bond stretches 2. Triatomic bond angle deformations 3. Tetra-atomic bond torsions 4. Van Der Waal's repulsion 5. Electrostatic attractions of bond dipoles.
The MM will be used is MM2 force field calculation. To minimize the total energy of the molecular system, the optimised structure can be found. Since the method only take the energy and bond properties, the semi-empirical molecular orbital, or the electronic aspects of reactivity cannot be shown. MOPAC calculation method will be a choice, because it takes the electrons and hence the semi-empirical orbital into account. MOPAC has many sub-method can be used. Therefore, the two methods, MM2 and MOPAC will be used together to provide the behaviour of the molecule and accurate results.
The Hydrogenation of Cyclopentadiene Dimmer
Structure of Cyclopentadiene Dimmer
The formation of cyclopentadiene is a Diels-Alder reaction. At room temperature, the endo product will be the major one. MM is used to show that the endo product is a higher energy conformation than the exo product. Therefore the reaction is under kinetic control.


The following data is obtained by optimising the structure of the two molecules with MM2 from the ChemBio3D. It shows that the total energy of exo product is lower than that of endo product. Also if we look at the individual terms of the energy, the difference of torsion of two products gives the highest value, and the others are quite similar since the dimmer is a bend ring. It means mainly the energy difference is from the steric effect of the molecule.
Energy values of the two isomer by MM2 (kcal/mol)
| Interaction Types | exo | endo | Difference (exo-endo) |
| Stretch: | 1.284 | 1.251 | 0.033 |
| Bend: | 20.580 | 20.847 | −0.267 |
| Stretch-Bend: | −0.838 | −0.836 | −0.002 |
| Torsion: | 7.656 | 9.511 | −1.855 |
| Non−1,4 VDW: | −1.418 | −1.545 | 0.127 |
| 1,4 VDW: | 4.235 | 4.321 | −0.087 |
| Dipole/Dipole: | 0.378 | 0.448 | −0.070 |
| Total Energy: | 31.877 | 33.998 | −2.121 |
From the bond angle values, the 2 hydrogen atoms on the carbon-carbon bond newly formed for exo-isomer is 112°, and that of endo-isomer is 110°. The two hydrogen of endo-isomer are pushed together. Also, the carbon-carbon-carbon bond angle, which two carbon are from the newly formed bond and the third carbon is from the alkene of the dienophile, of exo-isomer is 114° and that of endo-isomer is 116°. So the ring planes of endo-isomer is more bend. As the results of above, the endo-isomer gives the higher energy of torsion.
Hydrogenation of the Endo Product
The following data is obtained by optimising the structure of the two molecules with MM2 from the ChemBio3D. Product 4 shows the lower energy and so it is the stable one. The bend energy gives the most amount of the difference of energy. This is from the bonding angle of the alkene. Generally, the sp2 hybridised centre is 120°. Product 3 has angle of 127.1°. Compare to product 4, 124.4° and 124.9°, the bend energy of product 4 is lower than 3. The origin endo molecule has a higher bend energy than both of the product 3 and 4. It means the bend interaction energy of the two alkenes are different. Hydrogenation of the alkene near the carbon bridge can lower the bend energy more. So the product 4 is the major thermally favoured product.


| Interaction Types | Product 3 | Product 4 | Difference ((3)−(4)) |
| Stretch: | 1.2346 | 1.0969 | 0.138 |
| Bend: | 18.9428 | 14.5245 | 4.418 |
| Stretch-Bend: | −0.761 | −0.5492 | −0.212 |
| Torsion: | 12.1177 | 12.4979 | −0.380 |
| Non−1,4 VDW: | −1.4989 | −1.0702 | −0.429 |
| 1,4 VDW: | 5.7284 | 4.5115 | 1.217 |
| Dipole/Dipole: | 0.1631 | 0.1406 | 0.023 |
| Total Energy: | 35.9266 | 31.152 | 4.775 |
Stereochemistry and Reactivity of an Intermediate in the Synthesis of Taxol
Introduction
Taxol is an important drug in the treatment of cancer. There are two possible intermediates in the synthesis of taxol. They show an example of atropisomerism, which is the distinction between two molecules due to the restricted rotation about a single bond. The reason for this can be a ring structure or steric effect. In this case, it is the C=O group cannot rotate freely.


The following data is obtained by optimising the structure of the two molecules with MM2 and MMFF94 from the ChemBio3D. Because of the cyclohexane ring in the two intermediate, there are two conformations for each intermediate, chair and twisted boat. The chair conformation has the lower energy in both intermediates and the energy of them will be illustrated and discussed. To obtain the correct minimum value of the intermediate, manual edit the structure of molecule is performed.
The energy of Intermediate 9 and 10 by MM2 and MMFF94
| Interaction Types | Intermediate 9 chair | Intermediate 10 chair | Difference ((3)-(4)) |
| Stretch: | 2.787 | 2.620 | 0.166 |
| Bend: | 16.540 | 11.345 | 5.196 |
| Stretch-Bend: | 0.431 | 0.344 | 0.087 |
| Torsion: | 18.254 | 19.666 | −1.412 |
| Non−1,4 VDW: | −1.556 | −2.162 | 0.606 |
| 1,4 VDW: | 13.108 | 12.872 | 0.236 |
| Dipole/Dipole: | −1.725 | −2.002 | 0.277 |
| Total MM2 Energy: | 47.840 | 42.683 | 5.157 |
| Total MMFF94 Energy: | 70.551 | 60.551 | 10.000 |
From the table, it can be seen that the energy of intermediate 10 is lower than that of intermediate 9 in both cases of MM2 and MMFF94 minimization. This indicates that the intermediate 10 is thermally favoured intermediate during the synthesis. The main energy difference is from the bend interaction. The bond angle value of the sp2 hybridised carbonyl group of intermediate 10 is 119° and that of intermediate 9 is 125°. Intermediate 10 has the lower bend interaction and the value is closed to the ideal. The MMFF94 method suggests higher values for both of the intermediates. But the intermediate 10 is still at lower energy.
The functionalisation of alkene group is slow. The reason for this is the bond angle of the sp2 hybridised is already 120°. If the hybridisation of the alkene is changed, the bend interation and torsion may increase since there is a cyclopentane ring nearby. Increased strain and steric effect will make the product higher energy. So the rate of functionalisation of alkene group is slow.
Regioselective Addition of Dichlorocarbene
Introduction
The reactivity of dichlorocarbene will be investigated using MOPAC which can perform quantum mechanism. The method is based on semi-empirical molecular orbital theory, so the molecular orbital of dichlorocarbene can be produced. With the help of this, the regioselectivity can be shown. There are two kinds of addition for alkene, syn and anti. The major one will be determined by molecular orbital.

Part 1 Molecular orbital and reactivity
The molecule structure is optimised with MM2 first and then MOPAC/PM6. The results from MOPAC/PM6 method failed to generate symmetric molecular orbitals. Therefore MOPAC/RM1 is used.




From the orbital diagrams generated, it could be seen that the HOMO and HOMO-1 show syn structure. Alkene is relatively electron deficient so the electrons in HOMO have the highest energy and interact with the incoming electrophile, since the addition of dichlorocarbene undergo electrophilic addition as noted. The LUMO +1 shows an anti orbital. Since it is unoccupied, it is not favoured by electrophilic addition. The LUMO looks like the C-Cl anti orbital. Because of the electronegativity of Cl, the electron density of adjacent C will be drain away. HOMO-1 is the syn occupied orbital at the opposite side of Cl. The diagram shows HOMO-1 is close to the electron deficient carbon, so it will lose the electron density. As the result, dichlorocarbene will undergo syn addition at the alkene located at the same side of Cl.
Part 2 Influence of the Cl-C Bond on Vibrational Frequencies of Dichlorocarbene
The IR spectra for the two alkenes are generated from the SCAN. The structures of the two molecules are optimised with MM2 and then MOPAC/PM6. The results are used to create the files sent to SCAN.


The C-Cl bond frequency of monoalkene has a higher value than that of dialkene. The reason for this is the HOMO-1 C=C bond orbital is replaced by C-C bond, so there is no syn π bonding orbital, and the electron orbital shape changes. C-Cl bond is farther away from the new orbital and cannot drain electron density, hence becomes unstable. As the result, the bond will be at a high energy level and show a higher vibration frequency.
The vibration frequency of C-Cl bond in dichlorocarbene is 770.91cm-1. The frequencies of two C=C bonds are 1757.34cm-1 for HOMO and 1737.03cm-1 for HOMO-1. In monohydrogenated dichlorocarbene, the vibration frequency of C-Cl is 774.95cm-1. The frequency of the C=C bond is 1758.05cm-1.
Monosaccharide chemistry: Glycosidation
Introduction
Glycosidation is a substitution on the monosaccharide ring. It is group X is substituted by a nucleophile. The axial and equatorial products can be controlled by the orientation of the OAc neighbouring group. The oxonium cation can under go intramolecule reaction involving OAc group to form the intermediates. The intermediates can block a face from the attack of nucleophile to give different anomer. This is an example of neighbouring-group-participation.


Results and Discusstion
The total energy of the oxonium cation and intermediate A, B, C, D and their ring flipping conformation (A’, B’, C’, D’) are calculated with MM2 and MOPAC. Since MM2 method only take the five bond interactions mentioned above into account, the effect of charge on the molecule will be missed. While MOPAC uses quantum mechanism, so its results will be more reliable.
The R group used here is methyl group. This is the simplest group except H atom and MeOR is not polar. So there is no additional strong dipole-dipole interaction or H bond in OH group. The energy of the ring structure will be affect little by the side groups.
The MM2 and MOPAC/PM6 energies of products (kcal/mol)
| Interaction Types | A | A' | B | B' | C | C' | D | D' |
| Stretch: | 2.228 | 2.286 | 2.437 | 2.427 | 2.083 | 1.794 | 1.834 | 2.650 |
| Bend: | 11.585 | 14.379 | 12.742 | 14.248 | 14.368 | 16.338 | 21.591 | 20.087 |
| Stretch−Bend: | 0.828 | 0.967 | 0.946 | 0.926 | 0.777 | 0.704 | 0.876 | 0.853 |
| Torsion: | 2.947 | 2.548 | −0.011 | 0.954 | 9.447 | 7.408 | 6.947 | 7.485 |
| Non−1,4 VDW: | −2.237 | −2.546 | −0.260 | −1.120 | −2.285 | −4.307 | −1.428 | −3.310 |
| 1,4 VDW: | 20.001 | 18.885 | 18.768 | 18.868 | 17.646 | 17.482 | 16.371 | 18.999 |
| Charge/Dipole | −20.216 | −15.300 | −22.428 | −11.727 | −7.467 | −4.403 | −12.860 | −7.225 |
| Dipole/Dipole: | 6.876 | 9.895 | 7.102 | 7.474 | −2.059 | −0.623 | −0.105 | −0.369 |
| Total MM2 Energy: | 22.012 | 31.114 | 19.296 | 32.049 | 32.509 | 34.392 | 33.226 | 39.170 |
| MOPAC/PM6 Heat of Formation Energy: | −91.176 | −75.907 | −88.525 | −68.234 | −91.661 | −91.661 | −84.078 | −66.504 |
From the total energy results, the two methods, MM2 and MOPAC/PM6 are agreed to some extent. Both of them suggest that the origin conformations, A, B, C and D, have the lower amount of energy and are more stable, except C, which has the same amount of energy with its ring flipping conformer. The difference need to be noticed is MM2 results show oxonium cation A has more energy than B, while MOPAC shows reverse results. The difference is from the mechanism used by the two methods, so they will use different parameter to calculate the results. During the calculation of MM2 results, Chemdraw3D will say some parameter is guessed. However, MOPAC uses quantum mechanism which will give accurate calculation.
The energy differences between the flipping conformers are mainly from the dipole/charge interaction by looking at MM2 calculation. The positive charge on the six-membered ring oxygen will interact with the dipole on the neighbouring acyl group oxygen. The distance between the charge and dipole is responsible for the energy difference. Also, bend interaction, from the bond angle, and dipole-dipole interaction, affected by the axial and equatorial position, are quite take large ratio in the energy difference.
The energy results from MOPAC suggest that A and C have the similar energy, and so are B and D. The results mean before the formation of bond, the cations are as stable as each other. So A and C/ B and D must have very similar structure and the atom distance, angle and orientation of acyl group will be same. A/B is ready to form a bond to turn into B/D. Each of A’/C’ and B’/D’ are not at the same energy since the flipped structure change the axial and equatorial positon, hence different atom distances between non-bonding atoms. So energy is not same.
Compare the energy of C/C’ pair and D/D’ pair, C/C’ may show a weak diastereoselectivty since the MOPAC energy of the two ions and same, though MM2 shows C is at lower energy. The route via C’ conformer will be possible. D/D’ will show a strong diastereoselectivty since the energy difference between the ring flipping conformer and the origin one is big. Only the route shown in introduction is chosen during glycosidation.
Mini Project Assigning regioisomers in "Click Chemistry"
Introduction
Click Chemistry describes the chemical reactions generating products fast by joining small units together. It is a description for the chemical reactions which give high yield, inert byproduct, and show high stereoselectivity with good atom economy. The reaction driven force is big, the products are thermal favoured.
The 1,3-dipolar cycloaddition between an azide and an alkyne to give a 1,2,3-triazole is an example of it. The products of this reaction gives two isomer, 1, 4-isomer, which is the major product under Cu (I) catalyst, and 1,5-isomer, which is the major product under Ru (II) catalyst. The reaction can take place at room temperature and the yield is very high. The computational technique will be used to predict the spectra of the products and tell the difference between them. Compare with the literature, the accuracy of computational methods will be found.
The specific pair of molecules chosen are R1 = -C(CH3)2OH and R2 = -CH2C6H5. The reason for this is the selectivity of one isomer is very high (>90%) under certain catalyst, also the total number of non-carbon atoms is small.
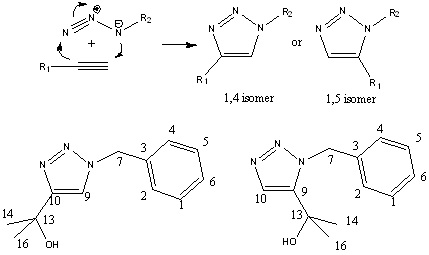

The structure of the two products at minimum energy
The structure of 1,4-isomer and 1,5-isomer are optimized with MM2 method and then MOPAC/PM6 methods. The energy results are illustrated below.
Minimum energies of 1,4 isomer and 1,5 isomer by MM2 and MOPAC/PM6(kcal/mol)
| Type of Interactions | 1,4 isomer | 1,5 isomer | Difference (1,4 isomer-1,5 isomer) |
| Stretch | 0.562 | 0.710 | −0.148 |
| Bend | 13.798 | 14.092 | −0.294 |
| Stretch−Bend | 0.018 | 0.073 | −0.055 |
| Torsion | −5.628 | −5.051 | −0.577 |
| Non−1,4 VDW | −5.179 | −4.583 | −0.596 |
| 1,4 VDW | 9.059 | 9.582 | −0.523 |
| Dipole/Dipole | −0.992 | −1.048 | 0.056 |
| MM2 Energy | 11.639 | 13.775 | −2.136 |
| MOPAC/PM6 Heat of Formation | 19.149 | 18.963 | 0.186 |
The total energy from MM2 and MOPAC/PM6 give similar values. The difference of minimum energy is small, though MM2 suggests 1,4-isomer is at a lower energy state, while MOPAC suggests 1,5-isomer is more stable. So if the reaction takes place directly, both of the products may be formed with similar ratio. The value of minimum energy is small so the reaction can take place at room temperature with a fast rate. Both of the products are similarly thermal favored. It also suggests the reason for high selectivity under different catalysts. The catalyst increase the rate of producing one of the product, and the major product will be the kinetic favored.
13C NMR spectra
The structure of 1,4-isomer and 1,5-isomer are optimized in ChemBio 3D with MM2 method. The geometry is then optimized with DFT by using SCAN. The output results is used to calculated the NMR spectra by using SCAN again.


NMR peak values of 1,4 isomer and 1,5 isomer
| Environment | Chemical Shift/ppm | Literature | Difference | Environment | Chemical Shift/ppm |
| 14 | 30.97 | 30.9 | 0.07 | 14 | 31.48 |
| 16 | 32.03 | 30.9 | 1.13 | 16 | 30.47 |
| 7 | 53.44 | 52.94 | 0.5 | 7 | 54.71 |
| 13 | 68.84 | 67.63 | 1.21 | 13 | 69.7 |
| 6 | 124 | 127.82 | −3.82 | ||
| 2 and 4 | 124.17 | 128.15 | −3.98 | 1, 2, 4 and 6 | 124.58 |
| 1 | 124.72 | 128.91 | −4.19 | ||
| 5 | 125.4 | 130.58 | 5.18 | 5 | 125.38 |
| 10 | 128.14 | 130.66 | −2.52 | 10 | 154.83 |
| 3 | 134.24 | 136.62 | −2.38 | 3 | 133.33 |
| 9 | 138.12 | 144.16 | −6.04 | 9 | 117.17 |
The literature values of 1,5 isomer are compared with the values from computational method. The value differences are quite small, except the chemical shift of ring carbon and C(9). Compare the chemical shift of the two molecules, the main difference is at the C(9) and C(10). They are at the different environments in the two molecules. In 1,4 isomer, C(10) is near the side group so it is more deshielded, and gives a higher value. C(9) gives a lower value. In 1,5 isomer, C(9) is near the side group.
Overall, the computational method gives a close and accurate result comparing with the results from laboratory. It also correctly assigned the peaks. If a mixture of the two product are formed, the resultant spectrum should show pair of C(9) and C(10).
IR Spectra
The output results of geometry are used to calculate the IR spectra by using SCAN. It is the same data for NMR calculation.


Bond vibration values (cm-1)
| Bond | 1,4 isomer | 1,5 isomer |
| C-N | 1207 | 1142 |
| C=C | 1502 | 1502 |
The spectra shows that the C-N bond absorption of 1,5 isomer has a lower value than that of 1,4 isomer. The benzyl group in 1,5 isomer is close to the C(CH3)2OH group. The steric effect will make the two groups repel each other, so the C-N bond length is increased. The bond is weaken, and show a smaller value on the IR spectrum. The alkene C=C of the two molecules have the same value, means it is not affected by the steric effect too much.
Conclusion
The computational methods are accurate to calculate the minimum energy structure of molecules, the bond lengthes and bond angles. From these values, the reactivity of the molecules and the reaction can take place will be predicted. For more complex calculations like generating NMR and IR spectra, the accurate results can be obtained by using advanced and sophisticated methods. So in the future, the computational technique will be a important tool to determine and predict the reaction product, especially with stereoselectivity.