
Rep:Mod:XYZ456
Jeffrey Gorman: Experiment 1C
Conformation Analysis using Molecular Dynamics

Dimerisation of Cyclopentadiene
| Total Energy | (1) (kcal/mol) | (2) (kcal/mol) |
|---|---|---|
| Overall | 55.37346 | 58.19076 |
| Electrostatic | 13.01371 | 14.18410 |
| van Der Waals | 12.80120 | 12.35590 |
| Torsional | -2.73070 | -2.94945 |
| Angle Bending | 30.77286 | 33.19323 |
Cyclopentadiene dimerises to produce the kinetic product (2) rather than the more thermodynamically stable (1). This is reflected in the molecular modelling and MMFF94(s) optimization of the two products. The total energies of (1) and (2) are 55.37346 kcal/mol and 58.19076 kcal/mol respectively. Therefore (1) is more stable. In the energy comparison graph above there is little deviation between (1) and (2) in the values provided. In general however energy values for (1) are closer to 0 kcal/mol (bond ideality) and so overall total energy is lower and more stable.The dimerisation occurs vi a bis-pericyclic reaction. Under heat/thermodynamic conditions the cycloaddition 4n+2 electron suprafacially with disrotation; meaning the transition state has a plane of symmetry and forms via Huckel aromtricity with no twist in the aromatricity.[1] Upon visual inspection of the optimized dimers, we would expect steric repulsion between the bridging-methyl hydrogens and ring hydrogens (highlighted in red below) in the exo product which would cause greater angle and bond strain in the molecule. van Der Waals energies suggest that there is not a substantial difference between the isomers, however angle bending for (2) is greater suggesting greater angle strain in the exo molecule.
| (1) | (2) |
|---|---|

|

|
However (2) is the major product of the dimerisation, thus we can conclude the reaction forms the faster-kinetic product, going by kinetic conditions under an irreversible reaction; because of its early, low-energy transition state with a lower Eact.The endo product is often expected in Deils-Alder reactions due to the secondary orbital interactions of the two molecules, in this case the two cyclopentadienes, which give the transition state stability. The exo product, despite being thermodynamically stable, does not have transition state stabilitsation of secondary orbital interactions and therefore has an earlier (and higher energy) transition state. If this reaction were under equilibrium conditions we expect the more thermo-stable (1) molecule to be the major product.
Hydrogenation of the Cyclopentadiene Dimer
| (3) | (4) | ||||||
|---|---|---|---|---|---|---|---|
|
|
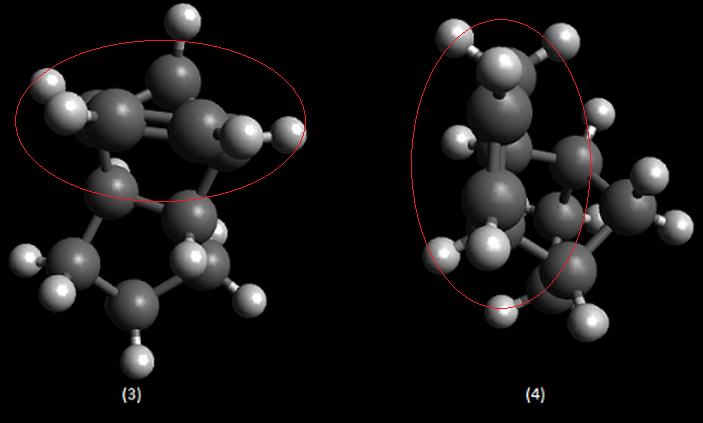
Upon completing optimizations of the hydrogenation products (3) and (4) yielded total energies of 50.44574 kcal/mol and 41.25749 kcal/mol respectively. (4) is the more stable regioisomer as it lacks a double bond and bridging-group converging on the same carbon, significantly reducing bond/angle strain in the molecule. This lack of strain is seen in both the torsional and stretch bending total energies below; where both values deviate from 0 kcal/mol more. Inspection of the olefin hydrogens shown in (4) shows the surrounding hydrogens form a staggered conformer in the Newman project, reducing repulsion. However in the regioisomer (3) the olefin hydrogens and surrounding ring carbons form an eclipse-like conform.
{kind=link}
| Total Energy | (3) (kcal/mol) | (4) (kcal/mol) |
|---|---|---|
| Overall | 50.44574 | 41.25749 |
| Torsional | -1.50151 | -0.37847 |
| Stretch Bending | -2.10450 | -1.65718 |
| Electrostatic | 13.01371 | 14.18410 |
| Angle Bending | 5.11942 | 5.14702 |
| van Der Waals | 13.63742 | 10.63743 |
More negative stretch-bending in (3) suggests bonds in the molecule were unable to stretch compared to an ideal carbon-carbon sigma bond, suggesting bond strain; the larger angle bending in (4) suggests less angle strain. Both observations coincide with our previous suggestion of an olefin and bridging methyl groups causing strain in (3). (3) has greater van Der Waals forces within the structure, which will lower thermodynamic stability further. The magnitude of the electrostatic energy is as we expect. (3) has the lower (more zero-like) energy for electronics. This suggests more stable electronics and may well be an attributing factor to the kinetic preference of the reaction. Thus (3) has the lowest electronic/kinetic pathway.
Energy Required to Form (3) from (2) = -7.74502 kcal/mol
Energy Required to Form (4) from (2) = -16.9333 kcal/mol
Values for the above were calculated via subtraction against (2) total overall energy found above. Under the optimizations undertaken (4) is significantly more thermodynamically favorable than (3).(3) has more negative torsional energy and stretch-bend energies because of the unfavorable sp2 carbon being one bond away from an already strained bringing group; making the ring structure constrained. This makes the stretching modes in (3) more rigid than (4). We can therefore conclude that because (4) has a lower overall total energy it is the stable, thermodynamic product in the hydrogenation of a cyclopentadiene dimer. If we assume this catalyzed reaction is reversible, we can say in confidence that the reaction is under thermodynamic control and (4) is the major product.
Atopisomerism in an Intermediate to the Synthesis of Taxol
| (9) | (10) | ||||||
|---|---|---|---|---|---|---|---|
|
|
Above are the optimized (same optimizations as cyclopentadiene) taxol synthesis intermediates, the key difference being the ketone being above (9) and below (10) the bi-cyclic carbon framework. One noticeable difference between the two optimized structures is the conformation of the 6-membered cycle. In (10) the preferred conformation is the more eclipsed-like structure that resembles the twist-boat/boat-C6H6 conform. However (9) possess a more chair-like structure. For cyclohexane the chair is significantly more stable then the boat conform due to a lack of eclipsing hydrogen repulsion.
| Total Energy | (9) (kcal/mol) | (10) (kcal/mol) |
|---|---|---|
| Overall | 70.54687 | 66.37596 |
| Torsional | 0.12465 | 3.87212 |
| Stretch Bending | -0.07212 | -0.13827 |
| Electrostatic | 0.29752 | -0.04888 |
| Angle Bending | 28.33315 | 19.07896 |
| van Der Waals | 33.17873 | 34.89887 |
| Bond Stretching | 7.70030 | 7.74959 |
(10) has a more negative energy value and therefore is the more thermodynamically stable structure. This is contradictory considering the 6-membered ring taking the commonly less-stable twist-boat/boat-like conform. However they key difference between the two is from the angle bending. The greater positive energy for angle-bending in (9) suggests that the bond angles are far from natural lengths suggesting larger distortions from ideal bond angles in the molecule.
In both isomers the larger cyclic ring is constrained by the double bond and bridging carbon group. During optimization in (9) forcing the carbonyl to be in the same plane as the bridging -C(CH2)2 creates a more distorted ring structure making the molecule visually more compact. In the above jmol of (9) the 6-membered ring is considerably more endo-like. This attributes the (9) having larger angle bending energy and high van Der Waals interactions.
In the formation of the intermediate (10) will be the major product over (9). This alkene is known to react slowly in the preceding synthetic step. Maier and von Rague Schleyer have shown that multi-cyclic olefin molecules containing bridging are stabilized and unreactive.[2] This is conceptually surprising at first as when introducing significant angle/bond strain into a ring we expect the molecule to want to reduce this strain by bond-breaking/reacting. Under Bredt's Rule the molecule should be unstable. Localizing a double bond and bridgehead makes p-orbital overlap strained and difficult to achieve. This would make a pi-bond unstable to form at the bridgehead carbon as in both (9) and (10).[3]
However as the molecules are stable they must be anti-bredt molecules. The steric hindrance/crowding of the molecule may contribute to the molecule being relatively inert. But the overriding reason for stability is from the cage structure of the double bond and strain produced by the multi-cycloalkane. This is compounded with bridging molecules near the olefin which further add to the strain in the bi-cyclic molecule. Further to the work of Maier and von Rague Schleyer, Novak used computational models to prove a number of anti-bredt molecules were stable. The report suggests as in Maier and von Rague Schleyer's work that because of the ring/angle strain at the bridgehead/olefin carbon the double bond resembles that of a 1,2-diradical rather than a conventional double-bond.[4]
Olefins with bridging nearby, whilst caged in a multi-cyclic compound, are considered "hyperstable". The key reason for this stability, despite the bond strain, derives from the double bond being nearby a bridging group is thermodynamically more stable then the double bond being anywhere else in the molecule.[5] Whilst having bridgehead olefins in small rings does create instability/reactivity, larger and medium sized rings (>8 carbons) gain stability. Introducing double-bond character at the bridgehead carbon forces the bridging group to flatten, this flattening relives strain on the ring that had been introduced by the bridging group. Furthermore because the molecules are bicyclic systems also gain relief from the bridging group flattening. Reducing angle and bond strain. Kim found that the sp2 carbon is less sterically congested in a medium-sized bridgehead ring than an sp3 carbon because it receives less transannular/Prelog strain from hydrogens inside the same ring.[6] All of these factors go some way to explaining the inertness of taxol intermediates.
Spectroscopic Simulation using Quantum Mechanics
The following spectroscopic data was formulated for molecule (18)
1H-NMR
| (17) | (18) |
|---|---|
| Literature Shift (ppm)* | Literature Integration* | Computational Shift (ppm) | Computational Integration |
|---|---|---|---|
| 5.21 | 1 | 4.84 | 1 |
| 3.00-2.27 | 6 | 2.85-1.98 | 6 |
| 2.70-2.35 | 4 | 1.94-1.92 | 5 |
| 2.20-1.70 | 6 | 1.87-1.63 | 3 |
| 1.58 | 1 | 1.60-1.52 | 14 |
| 1.50-1.20 | 3 | 1.42-1.37 | 2 |
| 1.10 | 3 | 1.25 | 2 |
| 1.07 | 3 | 1.03 | 1 |
| 1.03 | 3 | 0.96 | 3 |
| 0.81 | 3 | ||
| 0.76 | 3 | ||
| 0.75 | 2 | ||
| Methyl averaging correction** = -15 | |||
| Total = 30 | Total = 30 |
Gaussview Computed Molecule (18) H-NMR Spectrum
{kind=link}
(*)Literature values of (18) taken in deuterated benzene[7]
(**)Gaussview calculates all 3 hydrogens in methyl groups separately rather than an averaged shift value with integral of three. This results in an incorrect number of hydrogens in the data. The 9 shifts were averaged to 3 shifts each with 3H integration and the incorrect additional integrals are removed from the data set with this correction factor.
Above is a tabulated comparison of the 1H-NMR of molecule (18), using experimental-literature values and computational methods. The approximations assumed in computational construction of the spectra are as follows:
- B3LYP theory. A hybrid function where the exchange function is combined with the exact Hartree-Fock theory. The Hartree-Fock theory assumes electronic and nuclear motions are independent. It further assumes relativistic effects can be ignored and further simplifications.
- 6-31G(d,p) basis. This basis set takes into account both hydrogen and carbon polarization.
Comparing the NMR data above we can see they general trend of chemical shifts and integration is visible. There is however a clear difference in chemical shifts overall, however only by 1-2ppm. This can be explained by the experimental values being performed at 200 MHz whereas the computed values are at an undefined frequency. As we expect, the approximations make the computational calculations limited. For example the basis set 6-31G(d,p) does not account for polarization of sulfur and oxygen in the molecule. The programs use Hartree-Fock approximations to make calculations significantly easier; however this will remove considerable accuracy from the values.
The most obvious difference is in the center of the spectra where computational analysis between 1.60-1.52ppm yields 15H integration. This is due to the similar shifts of a number of hydrogens which in the experimental NMR produced multiplets in that area. (N.B. it should be noted that the calculations yielded a number of separate singlets in this range, Gaussian does not give multiplicity, signals close to one another were combined in the table for simplicity.)
13C-NMR
| Literature Shift (ppm)* | Computational Shift |
|---|---|
| 211.49 | 212.14 |
| 148.72 | 148.55 |
| 120.90 | 119.26 |
| 74.61 | 119.36 |
| 60.53 | 89.08 |
| 60.53 | 68.21 |
| 51.30 | 56.64 |
| 50.94 | 56.39 |
| 45.53 | 50.44 |
| 43.28 | 48.00 |
| 40.82 | 45.68 |
| 38.73 | 42.76 |
| 36.78 | 39.06 |
| 35.47 | 37.60 |
| 30.84 | 33.37 |
| 30.00 | 30.92 |
| 25.56 | 30.20 |
| 25.35 | 27.76 |
| 22.21 | 26.84 |
| 21.39 | 25.11 |
| 19.83 | 23.39 |
Gaussview Computed Molecule (18) C-NMR Spectrum
{kind=link}
(*)Literature values of (18) taken in deuterated benzene[5]
Computed raw data for both C and H NMR[8]
The table above shows both literature and computational shifts in the 13C-NMR. Integration is ignored because of the low carbon-13 abundance. Compared to H-NMR, C-NMR is considerably more accurate to experimental values. (N.B. the same approximations were applied as in H-NMR computations). Both sets of results are carried out under deuterated benzene solvent and literature-experimental values at 75MHz.
Considering the accuracy of the computed and experimental carbon NMR and trend similarities of hydrogen NMR, molecule (18) appears to have the carbon-scaffold structure of the molecule correctly assigned and characterized. The trends of the H-NMR spectra are similar, however the shifts of all signals in the spectrum deviate by about 0.5ppm. This does call in to question whether the structure identified by Paquette et al. is correct. Analysis by Lodewyk et al. showed the small deviation in chemical shift of reported aquatolide was because the NMR had been assigned slightly wrong and the molecule was stable at a different conformation then had previously been understood.[9] The difference in computed and experimental spectra is not as great as discovered by Lodewyk, so the erroneous results could lie in the computational method. Unfortunately for molecule (18), no other NMR assignment in benzene solvent has been made so comparisons are unlikely at this point. However further calculations and comparison should be made (where programs/computations permit) with optical activity/rotation.
| Molecule | Gibbs Free Energy* (J/mol) |
|---|---|
| 17 | -1651.375742 |
| 18 | -1651.349575 |
(*)via Gaussview FREQ keyword. Calculated as "Sum of electronic and thermal Energies"
Above is the Gibbs Free Energy values for the two molecules (17) and (18). The values show the molecules are stable due to the negative Gibbs energies however there is very little thermodynamic difference between the two molecules. Clearly the introduction of the dithioacetal on the 6-membered ring has altered the stability of the two isomers (17) and (18); compared to their precursors (9) and (10). Previously (10), with the carbonyl pointing below the ring-plane, was considerably more thermo-stable then (9), where the carbonyl pointed above the plane. However addition of new groups in (17) and (18) have made their thermodynamic stability equal.
Analysis of the Properties of the Synthesised Alkene Epoxides
Analysis of Shi Catalyst

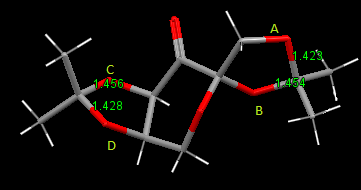
The crystal structure of the stable intermediate, formed during Shi catalyst synthesis (21), was computed from the original paper (by Shi et al.); using the Cambridge Crystal Database under Conquest and Mercury programs.[10] Bond lengths of the C-O bonds about the acetal groups in the molecule were calculated and labelled A-D. The bond lengths of which are recorded below:
{kind=link}
| C-O Bond Label | Bond Length (Å) |
|---|---|
| A | 1.423 |
| B | 1.454 |
| C | 1.456 |
| D | 1.428 |
The ketone and oxane in the central 6-membered ring will induce inductive effects on A-D. Both C and B are one carbon nearer to the ketone and both show a larger effect via induction due to their longer bond lengths. In order to assess the C-O bond length further, and by extension bond order/strength of each anomeric centre, semi-qualitative Newman projections through each bond were visualized in order to rationalize steric/environmental aspects effecting A-D. Further investigation involved assessing the natural bonding orbitals (NBO) of A-D and assess what anti-periplanar bonds were available and how they would affect bond length. Short contact lengths were also taken into account for the crystal structure in order to assess how intramolecular bond structure would affect the atoms/bonds involved. Whilst van Der Waals forces could not be calculated at this time, bond distances such as the two negative oxygen atoms in each ring were analysed to assess the dispersion forces within the ring.
A has the shortest A(C-O) bond. The groups in the Newman projection are not eclipsed-reducing Pauli repulsion and strain in the ring. A also benefits from the stabilization of E2 energy between the C-O and C-H bonds. 6 σC-O/σ*CH anti-periplanar interactions from the two CH3 groups. The oxygen in A(C-O) undergoes two short-contact interactions with a neighboring intermediate in the crystal structure. The two short-contact intramolecular interactions are smaller than the sum of the van Der Waals radii. This additional interaction likely reduces the bond order of the A(C-O) bond somewhat.
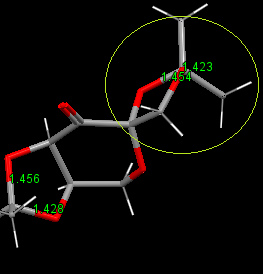{kind=link}
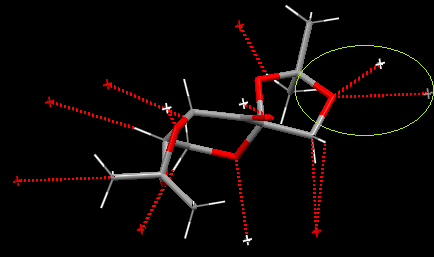{kind=link}
D has the next shortest C-O bond. Whilst its Newman projection shows significant unfavorable eclipsing, it has 6 σC-O/σ*CH anti-periplanar interactions from the two CH3 groups. The eclipsing will increase the steric and electronic repulsion of the surrounding groups. Stabilization of favorable anti-periplanar interactions and significant puckling in the ring to reduce oxygen-oxygen repulsion reduces the strain on the bond. The oxygen in this C-O bond does not have short contact interactions.
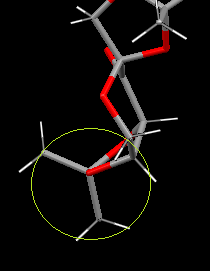{kind=link}
Looking through the B(C-O) bond we can see the atoms/groups are not eclipsed, the oxygen's (red) are reasonable far apart, due to the puckling of the 5-membered ring, as reduce bond strain and repulsion of the two occupying atoms. The C-O bond also gains stability from 8 favorable σC-O/σ*CH anti-periplanar interactions from the two CH3 and CH2 groups. The key difference between B and D is the larger distance between the electronegative oxygen atoms the the 5-membered ring holding D(C-O). The oxygen in this C-O bond does not have short contact interactions.
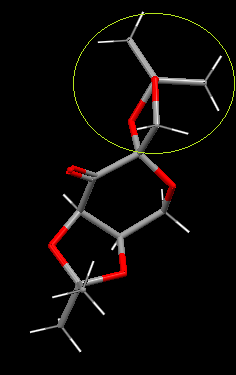{kind=link}
{kind=link}
C(C-O) has the longest bond. Looking down the C-O bond, surrounding groups are eclipsed so there are increases Pauli repulsions about the bond making it weak. C has 6 σC-O/σ*CH anti-periplanar interactions from the two CH3 groups. C has one short contact interactions from an adjacent Shi intermediate in the crystal structure. This spread of donation of electron density into intramolecular bonding will reduce the electron density on oxygen and by extension the C-O bond.
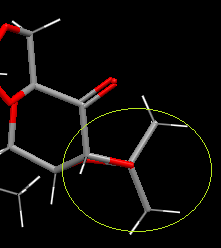{kind=link}
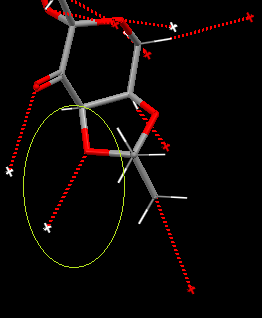{kind=link}
Analysis of Jacobsen Intermediate

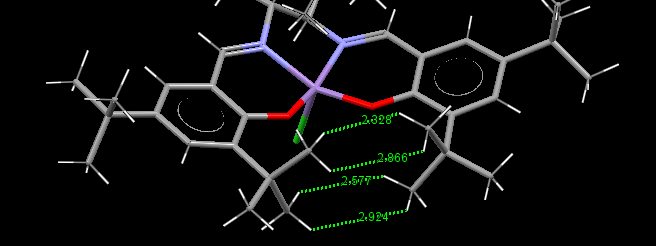
The distance between tButyl groups on the rings in the Jacobsen catalyst intermediate (23) is shown. H-H distances shown are greater than 2Å therefore we expect the dispersion forces here to be attractive.[11] This gives some favourable interactions to the molecule, there are also favourable dispersion forces between the two tButyl in each aryl ring.
{kind=link}
The distance is greater than the van Der Waals radius of any atoms involved however the steric bulk of the groups makes the vacant sight of the Manganese complex difficult to reach for any medium/large sized molecule from this end. This is further supported by the lack of short-contact sights for the crystal structure to intramolecularly bind. The steric hindrance of one of the Mn vacant sights is in agreement with the mechanism for the final Jacobsen Catalyst. In order to produce enantioselective epoxides the mechanism for alkenes binding to the catalyst needs to be incredibly specific.[12] The mechanism suggested by Katsuki bases the selectivity of the catalyst on the hinderance around the complex; and although this is not the final catalyst, the general form is already there in (23). Katsuki describes the approach of the alkene to the catalyst as coming from a specific side. The alkenes avoid the tert-Butyl areas of the aryl ligands and approach from behind, either side of the cyclohexane ring.
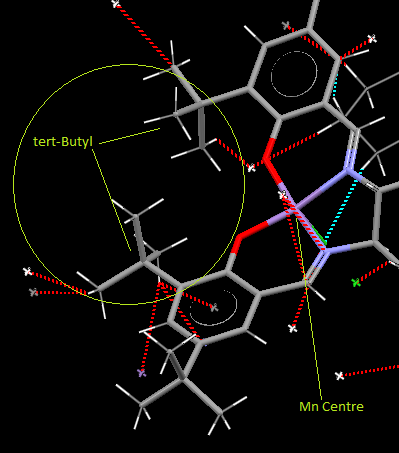{kind=link}
NMR Analysis of Epoxides and Epoxidation Transition State Analysis
| R-Styrene Oxide (29) | (R,R)-trans-Stilbene Oxide (30) | ||||||
|---|---|---|---|---|---|---|---|
|
|
R-Styrene Oxide (29)
1H-NMR
| Literature Shift (ppm)* | Computational Shift (ppm) |
|---|---|
| 7.28 | 7.39** (5H) |
| 3.817 | 3.66 (1H) |
| 3.091 | 3.12 (1H) |
| 2.758 | 2.54 (1H) |
Gaussview Computed Styrene Oxide H-NMR Spectrum
{kind=link}
Solvent=CDCl3
(**) These data points formed from the averaging of two very similar signals[13]
13C-NMR
| Literature Shift (ppm)* | Computational Shift (ppm) |
|---|---|
| 137.68 | 135.13 |
| 128.47 | 124.13 |
| 128.13 | 123.41 |
| 125.49 | 121.39** |
| 52.26 | 54.06 |
| 51.04 | 53.47 |
Gaussview Computed Styrene Oxide C-NMR Spectrum
{kind=link}
Solvent=CDCl3
Comparing data from both computational and experimental/literature methods shows very similar signals/shifts. This favorable comparison of chemical shifts suggests the minimized, computed conformation of styrene oxide is correct and matches how the molecule orientates itself in real life. Further analysis is required to characterize the stereochemistry of the epoxide. This can be achieved via computed optical rotation calculations about the bond. Jensen has shown that optical rotation (λ=589nm) yielded a rotation of -33.3o.[14] Computed optical rotation calculations via Gaussian using the same conformation as above was undertaken yielding: (λ=589nm)= -30.30 deg and (λ=365nm)= -94.60 deg.[15] The similarity of optical rotation of -30 degrees at 589nm confirms that the computed and experimental confirmations are effectively identification.
(R, R)-trans-Stilbene Oxide (30)
1H-NMR
| Literature Shift (ppm)* | Computational Shift (ppm) |
|---|---|
| 7.45-7.26 | 7.57-7.45 |
| 3.86 | 3.53 |
Solvent=CDCl3
(*) Literature values[16]
13C-NMR
| Literature Shift (ppm)* | Computational Shift (ppm) |
|---|---|
| 136.99 | 134.10 |
| 128.44 | 124.37 |
| 128.19 | 123.57 |
| 125.40 | 118.62 |
| 62.81 | 66.43 |
(*) Literature values are same as H-NMR
Computational data for C and H NMR[17]
As with styrene oxide, (R,R)-trans stilbene has very similar computed and experimental shifts in both carbon and proton NMR. This again suggests the optimized structure we have computed a very similar to the molecule that exists in reality. Further analysis of optical rotation needs to be carried out in order to assure the correct sterochemistry of the epoxide addition has been maintained. Computed calculation of optical rotation was performed yielding: (ʎ=589nm)= 298.14 deg. and (ʎ=365nm)= 1254.15 deg.[18] This value is confirmed by experimental literature values where Solladie-Cavallo et. al yielded rotations of 296 deg. at the same wavelength.[19]
Transition State Analysis for Shi Epoxidation of trans-Stilbene
| (R, R) transition (31) | (S, S) transition (32) | ||||||
|---|---|---|---|---|---|---|---|
|
|
Above are the two most stable transition states, from computed transitions, of the Shi epoxidation. Tables of all the available transition states and there energies can be found here. The formation of (R, R)-epoxide forms via attack at the re face whereas SS-epoxide is formed from the opposing se face, below the plane of the stilbene molecule. The oxygen with the least double-bond-character attacks the double bond in both transition states, and it is this oxygen that will form the epoxide in the alkene.
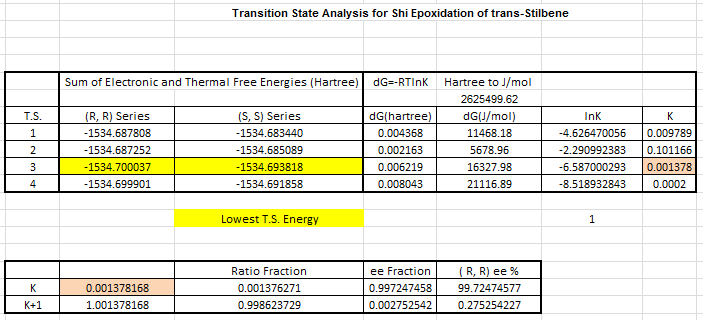{kind=link}

The most stable (R, R) transition gives an energy of -1534.700 Ha whilst the most thermodynamcially stable (S, S) transition is at -15.34.693 Ha. The difference in Gibbs Free Energy is calculated to be +16327.98 J/mol. As dG=-RTlnK, K (ratio of the two isomers) is found as 0.001378. This gives an enantiomeric excess of 99.72% in favor of (R, R) product. This value stands up to literature understandings of Shi catalyst specificity. Shi catalyst has been well documented as preferring trans alkenes for epoxidation, with very high selectivity. [20] Both transition states appear to show the spiro transition state rather then planar. The spiro transition state is theorized to be the lower energy, electronically favorable, transition state of the two.[21] The basis of this electronic stability is derived from the favorable orbital overlap of the oxygen lone pair in Shi catalyst and the π* orbital of the carbon-carbon double bond as shown to the right.[22]Further more the methyl groups at the both ends of the catalyst prevent a planar-like overlap due to repulsions of the relatively large groups, forcing the approach to be more spiro-like. This is identical to the transition states computed above and shown as Jmol files. Selectivity is increased by the attacked olefin being conjugated to an aryl group. In the case of trans stilbene there are two phenyl groups providing stabilization during the transition and therefore increased selectivity, hence our calculated ee value is very high, and yields very similar ee values (ca. 90%) with respect to (R, R)-stilbene epoxide.[23] Further analysis of the dispersion forces at play with regard to selectivity is displayed in section 3.5 where NCI/QTAIM analysis aids visually with description. The trans selectivity of the Shi catalyst can be understood by the interaction of favorable Hydrogen and dispersion forces/bonding between the catalyst and alkene. These interactions are more prominent in trans than cis as trans -alkenes are able to configure in a way where atoms in both molecules are nearer and can undergo non-covalent interactions. For the cis isomer much of this interaction is go as one R (aryl/alkyl, etc.) is oreintated in a different direction, increasing the distance between these favorable non-covalent interactions.
Transition State Analysis for Jacobsen Epoxidation of cis-β-methyl styrene
| (S, R) transition (41) | (R, S) transition (42) | ||||||
|---|---|---|---|---|---|---|---|
|
|

Above are the two most stable transiton states for the Jacobsen epoxidation. Both reactions occur on the same re face, however the difference in stereochemistry now derives from which side the of the complex the alkene approaches from. The phenyl ring is endo to the large planar conjugated aryl system about the central Mn atom. The oxygen atom that attacks the double bond and forms the epoxide is the axial oxygen that points directly upwards from the planar aryl ligands that occupy the equatorial positions about the metal centre of the complex. The alkene does not approach si to the oxygen as firstly the salen ligand blocks the path to the oxygen and is very repulsive and secondly the electronegativity of the chlorine atom will repel the electron-dense alkene pi-bond.
The most stable (S, R) transition state has an energy of -3383.258 Ha, whilst the most stable (R, S) transition station is at -3383.251 Ha. This difference gives a dG= 22314.123 J/mol and K= 0.000123. This results in a enantiomeric excess of (S, R) epoxide by 99.97%. Calculations and energy values of all transition states can be found here. This value agrees with research in the original paper reported by Jacobsen and Katsuki. The selectivity of Jacobsen epoxidations with cis-alkenes being in the excess of 90%.[10] cis-β-methyl styrene benefits from rather small, meaning it can more easily avoid the steric hindrance of the tert-Butyl groups on the salen ligand. As show in both by Linker et al. and the computed transition states above, the alkene approaches by (b). Both (a) and (b) are the most stable approaches, all others being blocked by the chloride or tert-Butyl groups. (b) is preferred due to the π-π interactions of the imine-bridge and olefin at the transition and more planar and less crowded environment compared to approaching above the cyclohexane ring. As previously stated, the alkene will not approach through the gap towards the complex between two tert-Butyl groups due to steric and electronic repulsive forces-making this method unfavorable. Jacobsen catalyst is selective towards cis alkenes as they are capable of approaching the oxygen atom without clashing significantly with protruding tert-Butyl groups. Trans configurations of the alkene will force the R groups around the carbon-carbon double bond to be nearer these large groups, increasing energy of the transition state and making it kinetically unfavored.
{kind=link}
Non-Covalent Interaction at Active-Site of Transition State for (R, R)-trans Stilbene
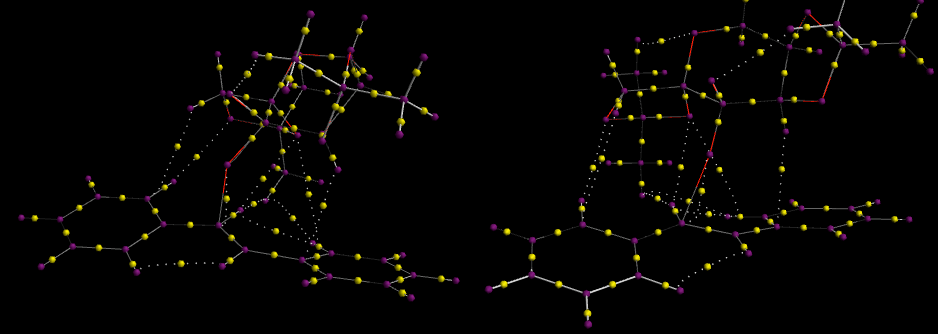
The NCI and QTAIM computations for (R, R)-trans Stilbene epoxidation transition via Shi catalysis, were computed for the more favorable thermodynamic and enantionmeric-excess transition state, as shown above in Jmol for molecule (31).
{kind=link}
Orbital |
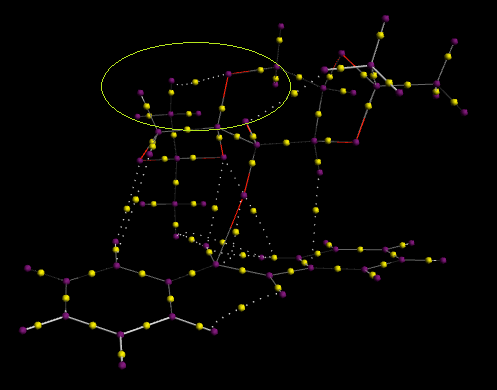
Above is the non-covalent interactions of the (R, R)-trans Stilbene epoxidation transition via Shi catalysis. The large band of green between stilbene and Shi catalyst suggests favorable mildly-attractive interactions about the area of bond forming (the blue ring). The major areas of interactions of this kind are localized about the now-broken double bond and ortho-hydrogens on the phenyl rings. QTAIM imaging of the molecule can be found here. Bond Critical Ponts circled in green show H-bonding between oxygen lone pairs on the catalyst and hydrogens on the alkene substrate. From visual analyses the BCPs appears to be nearer to the hydrogen of each of these bonds. Whilst the BCP circled in red involves an interaction of oxygen and carbon, this point is the site of epoxidation where oxygen will be transferred from the dioxirane group on the catalyst to the olefin. This is a bond about to be formed rather then a stabilizing interaction and is highlighted in the NCI Jmol above as a very attractive (blue) ring interaction. The BCP circled in blue shows non-covalent intramolecular interactions between an aromatic hydrogen and an allylic-like hydrogen in the saturated branch at the epoxidation site. This along with all other BCPs not mentioned are dispersion forces between non-polar carbon and hydrogen atoms, which gives an overall mildly attractive interaction between the two molecules-shown in red in the NCI Jmol above. Whilst Lane et al. have shown that BCPs are not necessarily correlated to H-bonding, they do give a good qualitative estimate to whether they exist or not.[24] Limitations of the QTAIM calculations do not give coordinates so effective analysis quantitatively is not possible at this point. It should also be noted in the NCI model, red, unfavorable interactions in the 5-membered shi rings results from electronic repulsion from the occupying acetal oxygens as previously theorized when considering minimizing conformation structure of the shi intermediate.
{kind=link}
These regions of interaction highlighted by green in NCI and bond critical points in QTAIM computations shows why the two molecules interact in this way. The beneficial H-bonding and dispersion forces can only occur on this side of the Shi catalyst. This occurs due to a number of factors. However computational modelling and visual inspection shows because the dioxirane forms at only one side, and because of the acetal-oxygens, favorable H-bonding occurs between the lone pairs on the oxygens and the hydrogens around the olefin site and on the substituted groups about it. The electron density at this side of the molecule create these favorable interactions and therefore alkenes approach/are-approached by the Shi catalyst selectively in this way. Furthermore when the alkene has a trans structure the interaction sites between the catalyst and substrate are larger/more numerous and this reduces the "bond/interaction" length for H-bonding and dispersion forces on both of the 5-membered rings in the Shi catalyst. If, for example, cis-stilbene is used, one phenyl ring will be pointing away from the catalyst. This will yield less non-covalent intermolecular interactions which would reduce the selectivity of the product formed. Alternatively in semi-qualitative terms, there will be less green, mildly attractive interactions between the two molecules with a cis-alkenes as one phenyl ring will be further away.
The structure/sterochemistry of the Shi catalyst therefore plays an important part in the NCI and enantioselectivity of the reaction. As previously shown when analyzing the structures of the Shi intermediate (21), the two methyl groups at each end of the Shi catalyst/intermediate create steric repulsion and hindrance to any incoming alkene approaching from the opposite side of the catalyst skeleton. Intramolecular H-bonding. may also be affecting the structure of the catalyst. It should also be mentioned that the spiro non-bonding orbital overlap between oxygen in the catalyst and the p-orbitals in the carbon-carbon double bond give further positive interactions during the transition state.
{kind=link}
Suggested Alkenes/Epoxides for Further Study

Pulegone/Pulegone epoxide is an reaction of note that shows some interesting optical rotation literature values. Reusch et. al has been the only person to assess the optical rotation of the epoxide at (λ=293 nm)= 786.5 deg and (λ=290 nm)= 786.5 deg.[25] The small change in wavelength to yield such a large change in rotation is in itself interesting, no further research or comparisons have been made on this relatively simple molecule. Pulegone epoxide gives a very large rotation and there are a number of stereochemical motifs that can be altered which will affect the optical rotation. For example the chiral centre in the ring, where a methyl and hydrogen is bonded. Also the stereochemistry about the two carbons the epoxide is bonded to. Pulegone is a naturally occurring molecule attained from oils in plants such as catnip.[26] Being a naturally occurring molecule in a relatively famous plant the molecule is widely available from companies such as Sigma Aldrich in both R and S forms. The relatively simple and small size of the molecule makes it ideal for fast computations and should make minimization of conformation easy. Whilst the alkene provides neither cis or trans stereochemistry, the importance of the carbonyl and the chirality of the methyl on the ring could well be the deciding factor in the enantioslectivity of Pulegone epoxide. As there is no cis/trans isomerism here the face (re or si) will be important and again the carbonyl and chiral center are vital to understanding the ee of Pulegone epoxide.
The toxicology of the molecule has been well documented, with initial interesting being if it was the contributing molecule to the behavioral affects of catnip to felines. Furthermore its toxicity to rats and insecticidic potency is of interest especially if these attributes are enantiomer specific. In folklore the epoxide has been used an abortifacient despite the levels required being lethal. This biological activity and how enantiomeric selectivity may affect this provokes further interest in this molecule.
Appendices
- All molecules computed in Part/Week 1 were drawn in ChemBio3D and exported to Avogadro. No optimizations/minimizing energy calculations were partaken in ChemBio3D. Minimization of energy were then completed in Avogadro under the MMFF94(s) force field as only simple elements (H, C, S, O) were used. For optimizations of geometry and minimization of energy the "conjugate gradients" algorithm was performed. Any further analysis was provided by Gaussview/HPC. These settings were applied to all molecules in week 1.
- Week 2 test page of advanced Jmol coding can be found here
References
- ↑ Pierluigi C., Paolo Q., and Lucio T., Unexpected Bispericyclic Transition Structure Leading to 4+2 and 2+4 Cycloadducts in the Endo Dimerization of Cyclopentadiene Journal of the American Chemical Society, 2002, 124, 7, 1130-1131
- ↑ W. F. Maier, P. Von Rague Schleyer, Evaluation and prediction of the stability of bridgehead olefins, J. Am. Chem. Soc., 1981, 103, 1893-1895
- ↑ Nelsen, S. F., et al Bredt's rule kinetically stabilized nitrogen-centered radical cations and radicals in the 9-azabicyclo[3.3.1]nonyl system. Journal of the American Chemical Society, 1980, 102, 702-707.
- ↑ Novak, I. Molecular Modeling of Anti-Bredt Compounds., Journal of Chemical Information and Modeling, 2005, 45, 334-338.
- ↑ Lalitha, S., et al. Cage geometry controlled hyperstability in bridgehead olefins., Tetrahedron Letters, 1990, 29, 4219-4222.
- ↑ J.S Kim, The Molecular Mechanics Evaluation of the Stability of Bridgehead Olefins Containing Medium Rings, Korean Chem. Soc., 1997, 5, 491-495
- ↑ L. Paquette, N. A. Pegg, D. Toops, G. D. Maynard, R. D. Rogers, J. Am. Chem. Soc.,, 1990, 112, 277-283.
- ↑ Jeffrey Gorman, Gaussian Job Archive for C20H30O2, 2013 DOI:10042/25992
- ↑ M. W. Lodewyk , C. Soldi , P. B. Jones, M. M. Olmstead , J. Rita , J. T. Shaw, and D. J. Tantillo, The Correct Structure of Aquatolide—Experimental Validation of a Theoretically-Predicted Structural Revision, J. Am. Chem. Soc., 2012, 134, 18550–18553.
- ↑ Zhi-Xian Wang,Susie M. Miller,Oren P. Anderson, and, and Yian Shi, Asymmetric Epoxidation by Chiral Ketones Derived from Carbocyclic Analogues of Fructose, The Journal of Organic Chemistry, 2001 66 (2), 521-530
- ↑ Manjeera Mantina, Adam C. Chamberlin, Rosendo Valero, Christopher J. Cramer, and Donald G. Truhlar, Consistent van der Waals Radii for the Whole Main Group The Journal of Physical Chemistry, 2009, 113 (19), 5806-5812
- ↑ Linker T., The Jacobsen-Katsuki Epoxidation and Its Controversial Mechanism, Angew Chem Int Ed Engl, 1997,36 (19), 2060-2062
- ↑ Jeffrey Gorman, Gaussian Job Archive for C14H12O1, 2013 DOI:10042/25983
- ↑ Frederick R. Jensen and Ronald C. Kiskis, Stereochemistry and mechanism of the photochemical and thermal insertion of oxygen into the carbon-cobalt bond of alkyl(pyridine)cobaloximes, Journal of the American Chemical Society, 1975 97 (20), 5825-5831
- ↑ Jeffrey Gorman, Gaussian ORP Job Archive for C14H12O1, 2013 DOI:[1] 10042/25990
- ↑ Organic Syntheses, Coll., 1998; 74, 91
- ↑ Jeffrey Gorman, Gaussian Job Archive for C14H12O1, 2013 DOI:10042/25983
- ↑ Jeffrey Gorman, Gaussian ORP Job Archive for C8H8O1, 2013 DOI:10042/25991
- ↑ Solladie-Cavallo, Arlette; Diep-Vohuule, Anh; Sunjic, Vitomir; Vinkovic, Vladimir, Tetrahedron: Asymmetry, 1996 , 7, 6 1783-1788
- ↑ Hongqi Tian,Xuegong She,Jiaxi Xu, and, and Yian Shi, Enantioselective Epoxidation of Terminal Olefins by Chiral Dioxirane, Organic Letters, 2001, 3 (12), 1929-1931
- ↑ K. N. Houk, Jian Liu, Nicholas C. DeMello, and Kevin R. Condroski, Transition States of Epoxidations: Diradical Character, Spiro Geometries, Transition State Flexibility, and the Origins of Stereoselectivity, J. Am. Chem. Soc., 1997, 119, 10147-10152
- ↑ Y. Shi, Organocatalytic Asymmetric Epoxidation of Olefins by Chiral Ketones, Accounts of Chemical Research, 2004, 37 (8), 488-496
- ↑ Tian, H., She, X., Shu, L., Yu, H., Shi, Y., J. Am. Chem. Soc., 2000, 122, 11551-11552
- ↑ Lane J. R., Contreras-García J., Piquemal J-P., Miller B., and Kjaergaard H., Are Bond Critical Points Really Critical for Hydrogen Bonding?, Journal of Chemical Theory and Computation, 2013, 9, 8, 3263-3266.
- ↑ William Reusch and Calvin Keith Johnson, The Pulegone Oxides, The Journal of Organic Chemistry 1963, 28, 10, 2557-2560
- ↑ Guenther E., The Essential Oils, D. Van Nordstrand, 1949, 575.