
Rep:Mod:WGW1993
Part 1 - Set Exercises
1 - Hydrogenation of the Cyclopentadiene Dimer
The dimerization of cyclopentadiene is a bispericyclic reaction, containing features of more than one type of pericyclic reaction.[1] The endo product 2 is formed exclusively during this reaction, though some isomerisation to the exo product 1 is observed at high temperatures.[2] Using a molecular mechanics approach, we aim to identify whether the dimerization reaction is thermodynamically or kinetically controlled. In addition, the partial hydrogenation of the endo product will be investigated by analysing the ground state energies of 3 and 4 to determine the ease with which each of the double bonds is hydrogenated.

Procedure and Theory
The MMFF94s force field in Avogadro was used to optimise the structures and hence find the lowest energies conformations. The total energy values generated are simply summations of a number of contributing energy terms: bond stretching (str) and bending (bnd), torsional strain (tor), van der Waals forces (vdw) and electrostatic interactions (es). These energy terms represent deviations from normality and therefore the dissection of the total energy into these contributions can provide specific information about the geometry of a particular molecule.
Comparing Compounds 1 and 2 - Thermodynamic or Kinetic Control?
The total energies of compounds 1 and 2 are as follows:
| Compound | Total Energy / kcal/mol |
|---|---|
| 1 | 55.37344 |
| 2 | 58.19070 |
It is clear from these data that the exo dimer is lower in energy and hence is the thermodynamically most stable product. The dimerization reaction must therefore proceed under kinetic control, because the endo product is formed in preference to the exo product.
Comparing Compounds 3 and 4 - Ease of Hydrogenation
Compounds 3 and 4 are the products of partially hydrogenating the endo dimer 2. The ease of hydrogenation of each of the alkene groups can be predicted thermodynamically by comparing the total energies of the products. The lower energy structure is more likely to form due to the greater thermodynamic driving force. The table below shows the total energies of each of the products and in addition includes the dissection of the total energies into the contributing terms. All energy terms are in units of kcal/mol.
| Compound | str | bnd | tor | vdw | es | Total Energy / kcal/mol |
|---|---|---|---|---|---|---|
| 3 | 3.30736 | 28.95217 | 0.06505 | 13.27727 | 5.12101 | 50.72283 |
| 4 | 2.82306 | 23.02856 | -0.37824 | 10.63709 | 5.14702 | 41.25749 |
Compound 4 has a significantly lower total energy than compound 3 and hence is predicted to be the major product under conditions of partial hydrogenation. Consequently, it can be deduced that carbon-carbon double bond remaining in compound 3 is more strained. This is confirmed by measurement of the bond angles for each of the alkene groups, with the sp2 carbons in product 3 having bond angles of 107.2 o compared to 112.0 o for sp2 carbons in 4. In addition, analysis of the individual energy terms indicates the differences in bond bending and van der Waals contacts are the most significant. The large positive bending term for 3 is due to the presence of the strained double bond. On the other hand, the van der Waals term is more difficult to explain. One possible reason relates to the envelope conformation of the saturated 5-membered ring that is present in the optimised structure of 3. The 5-membered ring bends below and underneath the 6-membered ring and could potentially lead to the formation of unfavourable van der Waals contacts.
2 - Atropisomerism in an Intermediate Related to the Synthesis of Taxol
Structures 9 and 10 are atropisomers of a key intermediate in the synthesis of the cancer chemotherapeutic drug Taxol. The synthesis of the intermediate, originally proposed by Paquette et al.,[3] involves a [3,3]-sigmatropic (oxy-Cope) rearrangement via an endo-chair transition state to initially give isomer 9, which can then change its conformation about the carbonyl group to give isomer 10. Paquette et al. found that the conversion from 9 to 10 was facile at room temperature, indicating isomer 10 is thermodynamically more stable. We aim to compare these experimental observations with calculations based on molecular mechanics.

Procedure
A similar procedure was used as in the previous exercise. The MMFF94s force-field option in Avogadro was first used to optimise the structures of the atropisomers before calculating their total energies. Manual editing of the molecular framework was also carried out in order to convert between the twist-boat and chair conformations of the fused cyclohexane group.
Determining the Most Stable Atropisomer - Twist-boat versus Chair Conformation
Upon optimising the structures of 9 and 10, two distinct conformers were found for each atropisomer: one with the fused cyclohexane in the chair conformation and the other with the ring in the higher energy twist-boat conformation. The following table displays the predicted energies for each of these 'local minima' and includes rotatable models for each structure.
| Compound | Total Energy / kcal/mol | |
|---|---|---|
| Twist-boat | Chair | |
| 9 | 77.90209 | 70.53964 |
| 10 | 66.28826 | 60.55152 |
The calculated energies show that overall the atropisomer 10 with the cyclohexane ring in the chair chonformation is the lowest energy structure. More generally, the structure of 10, with the carbonyl group on the opposite face to the bridging methylene, was predicted to be more stable compared to having the carbonyl on the same face. Having the carbonyl positioned on the less congested face leads to a reduction in the number of unfavourable steric interactions between the carbonyl oxygen and nearby hydrogen atoms. In addition, transannular interactions may also make an important contribution to the total energy, as noted by Elmore.[4]
Furthermore, both isomer 9 and 10 are more stable with the fused cyclohexane ring in the chair conformation. This is because the chair conformation is free from torsional strain whereas the twist-boat conformation is not, as in free cyclohexane.
Stability of the Bridgehead Alkene
Bridgehead alkenes, such as found in 9 and 10 are known to be very unreactive. They are so-called 'hyperstable alkenes'.[5] The remarkable stability is because the alkene is in fact less strained than the saturated bridgehead alkane. For example, the hydrogenated derivative of 10 in the chair conformation has a total energy of 69.53529 kcal/mol. The additional hydrogen atom is forced into an eclipsed geometry with a neighbouring hydrogen, causing a great deal of torsional strain. Consequently, there is predicted to be no thermodynamic force driving the functionalisation of the alkene, thus explaining the low observed reactivity.
3 - Spectroscopic Simulation of an Intermediate Related to the Synthesis of Taxol
Atropisomers 17 and 18 are functionalised derivatives of 9 and 10, differing only by a methyl and a 1,3-dithiolane group. As before, the reaction takes place at room temperature via an endo-chair transition state to form 17 with the carbonyl pointing 'up'. However, the additional steric bulk present in 17 results in elevated conformational barriers and means the product is in fact stable and isolable. The more stable atropisomer 18 forms after prolonged heating in THF.[6] Using quantum mechanics, we aim to predict the 1H and 13C NMR spectra for 17 and 18 and compare them with the experimental spectra of Paquette et al.[3]

Procedure
The structures of 17 and 18 were first optimised in Avogadro using the MMFF94s force field. The fused cyclohexane ring was positioned in the chair conformation for both atropisomers, as this was found to be the lowest energy geometry. Their NMR spectra were then simulated in Gaussian using the B3LYP DFT method, 6-31G(d,p) basis set, and deuterated chloroform as the solvent. The predicted shifts for the methyl groups were assigned and averaged in order to account for the degeneracy due to rapid C-C bond rotation. The deviations between experimental and simulated values across a range of shifts were calculated using the mid-point. The free energies of 17 and 18 were also predicted by computing the entropy and zero-point energy values.
NMR Spectra of 17 and 18 - Comparison with the Literature
Compound 17 NMR spectra: DOI:10042/25775
Compound 18 NMR spectra: DOI:10042/25776
1H NMR Spectra
Compound 17


| Calculated Shift / ppm | Experimental Shift / ppm | Integral | Deviation / ppm | Assignment |
|---|---|---|---|---|
| 0.82 | 0.80-1.00 | 1H | 0.08 | alkyl |
| 0.93 | 1.10 | 3H | 0.17 | methyl (c) |
| 1.11 | 1.25 | 3H | 0.14 | methyl (d) |
| 1.42 | 1.38 | 3H | 0.04 | methyl (g) |
| 1.31-2.82 | 1.35-2.80 | 14H | 0.01 | alkyl |
| 2.70 | 2.99 | 1H | 0.29 | alkyl (α-carbonyl, f) |
| 3.08-3.50 | 3.10-3.40 | 4H | 0.04 | dithiolane (k, l) |
| 5.86 | 4.84 | 1H | 1.02 | alkene |
Average Deviation = 0.22 ppm
There was good correlation between the calculated shifts and experimental literature shifts, with a low overall average deviation. However, although the shifts of the alkyl and dithiolane group protons were accurately predicted, there were two significant deviations that were observed. The first was the α-carbonyl protons, where the calculated shift was 0.29 ppm below the experimental shift. The second was the alkene proton, which had the largest observed deviation of 1.02 ppm.
Compound 18


| Calculated Shift / ppm | Experimental Shift / ppm | Integral | Deviation / ppm | Assignment |
|---|---|---|---|---|
| 1.17 | 1.03 | 3H | 0.14 | methyl (c) |
| 1.25 | 1.07 | 3H | 0.18 | methyl (d) |
| 1.41 | 1.10 | 3H | 0.31 | methyl (g) |
| 1.27-1.58 | 1.20-1.50 | 3H | 0.08 | alkyl |
| 1.58 | 1.58 | 1H | 0.00 | alkyl |
| 1.84-2.33 | 1.70-2.20 | 6H | 0.14 | alkyl |
| 2.47-2.65 | 2.35-2.70 | 4H | 0.03 | alkyl |
| 2.77-3.19 | 2.70-3.00 | 6H | 0.13 | dithiolane (k, l), alkyl |
| 5.98 | 5.21 | 1H | 0.77 | alkene (p) |
Average Deviation = 0.20 ppm
Again, there was good correlation between the theoretical and experimental shift values. As for 17, the alkene proton was predicted a much higher shift than the experimental value and was again the most significant deviation. In order to overcome this issue, a wide array of constrained alkenes could be modelled and their shifts calculated in order to deduce a reasonable correction which may be applied systematically.
13C NMR Spectra
Compound 17


| Calculated Shift / ppm | Experimental Shift / ppm | Deviation / ppm | Assignment |
|---|---|---|---|
| 22.52 | 18.71 | 3.81 | 6 |
| 23.76 | 20.96 | 2.80 | 12 |
| 26.25 | 23.86 | 2.39 | 4 |
| 26.86 | 25.66 | 1.20 | 1 |
| 30.56 | 26.88 | 3.68 | 2 |
| 32.25 | 28.28 | 3.97 | 18 |
| 36.08 | 28.79 | 7.29 | 10 |
| 41.87 | 32.66 | 9.21 | 15 |
| 46.28 | 35.85 | 10.43 | 11 |
| 48.66 | 38.81 | 9.85 | 14 |
| 49.03 | 39.80 | 9.23 | 7 |
| 51.90 | 45.76 | 6.14 | 13 |
| 53.97 | 46.80 | 7.17 | 5 |
| 55.20 | 48.50 | 6.70 | 3 |
| 57.36 | 52.52 | 4.84 | 17 |
| 62.31 | 56.19 | 6.12 | 9 |
| 92.99 | 72.88 | 20.11 | 16 |
| 116.96 | 125.33 | 8.37 | 19 |
| 149.69 | 144.63 | 5.06 | 20 |
| 220.12 | 218.79 | 1.33 | 8 |
Average Deviation = 6.49 ppm
The simulated spectrum was in good agreement with the literature, with a low average shift deviation observed. The general trend for the calculated shifts was that they are almost all higher field than the literature shifts, indicating a systematic correction is required to fit the simulated data to experimental data. The largest deviations were observed for carbon atoms alpha to heteroatoms. In particular, carbon atoms 14, 15 and 16 in the dithiolane group (and hence alpha to sulfur) had particularly large deviations, most likely due to spin-orbit coupling errors. Again, another correction is required, and from the data it seems a reasonable correction would be to reduce the shift by 9-10 ppm per alpha-sulfur atom.
Compound 18


| Calculated Shift / ppm | Experimental Shift / ppm | Deviation / ppm | Assignment |
|---|---|---|---|
| 21.72 | 19.83 | 1.89 | 6 |
| 22.62 | 21.39 | 1.23 | 12 |
| 24.55 | 22.21 | 2.34 | 4 |
| 26.46 | 25.35 | 1.11 | 1 |
| 28.16 | 25.56 | 2.60 | 2 |
| 33.65 | 30.00 | 3.65 | 18 |
| 34.13 | 30.84 | 3.29 | 10 |
| 38.74 | 35.47 | 3.27 | 15 |
| 41.77 | 36.78 | 4.99 | 11 |
| 41.82 | 38.73 | 3.09 | 14 |
| 46.77 | 40.82 | 5.95 | 7 |
| 49.14 | 43.28 | 5.86 | 13 |
| 49.69 | 45.53 | 4.16 | 5 |
| 54.20 | 50.94 | 3.26 | 3 |
| 54.88 | 51.30 | 3.58 | 17 |
| 60.42 | 60.53 | 0.11 | 9 |
| 94.11 | 74.61 | 19.50 | 16 |
| 120.14 | 120.90 | 0.76 | 19 |
| 148.07 | 148.72 | 0.65 | 20 |
| 212.23 | 211.49 | 0.74 | 8 |
Average Deviation = 3.60 ppm
Again, agreement with the literature spectrum is good, and overall the average deviation for 18 was smaller than for 17. In this spectrum, carbon atoms 14 and 15 in the dithiolane group had much smaller deviations than were found in the spectrum for atropisomer 17, which is surprising considering their chemical environment is expected to be very similar in both structures. On the other hand, a similar significant deviation (ca. 20 ppm) was again observed for atom 16.
Free Energies of 17 and 18
ΔG (17) = -1651.440162 hartrees
ΔG (18) = -1651.463258 hartrees
ΔΔG = 0.023096 hartrees = 60.639 kJ/mol
The ground-state energy of atropisomer 18 is significantly lower than that of 17 as expected from experimental observations.
References
- ↑ P. Caramella, P. Quadrelli and L. Toma, J. Am. Chem. Soc., 2002, 124 (7), 1130-1131. DOI:10.1021/jaja016622h
- ↑ J. E. Baldwin, J. Org. Chem., 1966, 31 (8), 2441-2444. DOI:10.1021/jo01346a003
- ↑ 3.0 3.1 L. A. Paquette et al., J. Am. Chem. Soc., 1990, 112 (1), 277-283. DOI:10.1021/ja00157a043
- ↑ S. W. Elmore and L. A. Paquette, Tetrahedron Letters, 1991, 32 (3), 319-322. DOI:10.1016/S0040-4039(00)92617-0
- ↑ W. F. Maier and P. Von Rague Schleyer, J. Am. Chem. Soc., 1981, 103 (8), 1891-1900. DOI:10.1021/ja00398a003
- ↑ L. A. Paquette et al., J. Am. Chem. Soc., 1991, 113 (4), 1335-1344. DOI:10.1021/ja00004a040
Part 2 - Prediction of the Properties of the Synthesised Alkene Epoxides
The Catalytic Systems - Shi and Jacobsen
Shi Catalyst
The Shi catalyst is a derivative of D-fructose and is involved in the asymmetric epoxidation of alkenes. Species 21 is the stable pre-catalyst which is converted to the active catalyst 22 in situ with the addition of an oxidant such as peroxomonosulfate (HSO5-). During this reaction, the ketone in 21 is converted to a reactive dioxirane group which can then transfer an oxygen atom to the alkene enantioselectively. High enantiomeric excesses (ee's) have been observed for unfunctionalised trans and tri-substituted alkenes.[1]

Crystal Structure and Mechanism
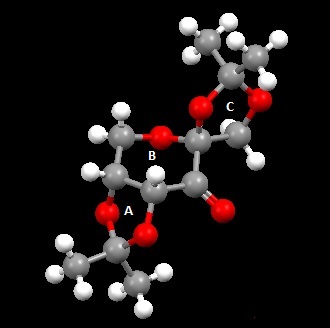
The crystal structure of 21 as determined by M. Ďurík et al. was analysed.[2] The table below shows the bond lengths of the O-C-O substructures in 21.

| C-O bond | Bond Length / Å |
|---|---|
| a | 1.415 |
| b | 1.422 |
| c | 1.455 |
| d | 1.422 |
| e | 1.456 |
| f | 1.428 |
The bond lengths a and b about the anomeric centre are similar in length, whereas the C-O bonds in the ketal groups (c and d, e and f) are significantly different. The enantioselectivity of the Shi catalyst is believed to arise from unfavourbale steric interactions present in the transition state between the dimethyl ketal groups on the catalyst and the substituents on the alkene, which results in large differences in energy for the different transition geometries.[3] For terminal alkenes, poor ee's are observed as steric congestion can be more easily avoided.[4] The asymmetry of the 5-membered rings A and C directs the ketal groups towards the reactive carbonyl group in space, leading to potentially greater interactions with alkene substituents in the substrate and hence causing the observed selectivity. In terms of ring conformations in the crystal structure , the central 6-memebered ring B has the chair conformation, while the 5-membered ring C has the envelope conformation. The other 5-memebered ring (A) has the higher energy half-chair conformation.
{kind=link}
Jacobsen Catalyst
The Jacobsen catalyst is a chiral (salen)manganese(III) organometallic complex. Species 23 is the stable precursor which is oxidised in situ using bleach (NaOCl) to form the active catalytic species 24 containing the metal carbonyl group. High ee's have been observed for unfunctionalised cis di-substituted, tri-substituted and conjugated alkenes.[5]

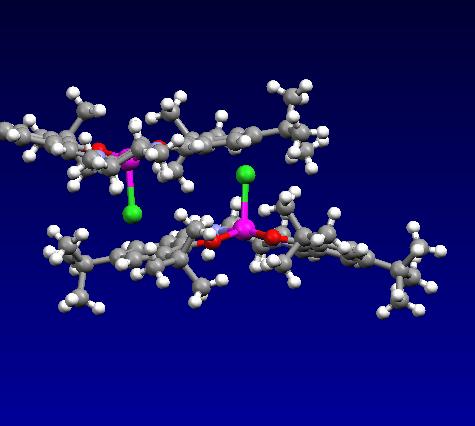
Crystal Structure and Mechanism
The crystal structure of 23 as determined J. W. Yoon et al. was analysed.[6] The most important feature of the molecule is the close approach of the the t-butyl groups ortho to the salen oxygens. These bulky groups disrupt the approach of an incoming alkene substrate from that side of the molecule by blocking access to the reactive metal carbonyl group. In addition, the t-butyl groups para to the salen oxygens obscure the approach across both of the aryl groups, and hence the lowest energy transition state involves approach of the substrate across the relatively unhindered cyclohexane ring. Enantioselectivity is driven by the stereochemistry at this position, whereby large substituents on the alkene are positioned away from the axial hydrogen pointing 'up'.[7] Trans di-substituted alkenes are found to have low yields and ee's as both possible transition state geometries have a substituent in close contact with this axial hydrogen.[8] Another intersting feature of the crystal structure is its curved surface, which presumably forms in order to ease the steric congestion of the ortho t-butyl groups.
{kind=link}
Alkenes Under Investigation - Styrene and trans-β-methylstyrene
The alkenes under investigation are styrene and trans-β-methylstyrene. Styrene is a terminal alkene and trans-β-methylstyrene is a trans di-substituted alkene. They can both undergo epoxidation to form the enantiomeric epoxide products, and under uncatalysed conditions would form a racemic mixture. The Shi and Jacobsen catalysts have the potential to produce an enantiomeric excess of one of the enantiomers over another by altering the energies of the possible transition states. In this section we aim to simulate the NMR spectra, optical rotations and transition state energies so that experimental products may be assigned absolute configuration. Non-covalent interactions and the electronic topology of a particular transition state system are also analysed in order to begin to explain the observed stereoselectivity.

Simulated NMR spectra of Epoxide Products
The same procedure was used to generate the NMR spectra for the epoxide products as was used for the Taxol intermediates 17 and 18. In addition, the couplings were also predicted for the 1H NMR spectra, using the B3LYP density functional method and the 6-311 G(d,p) basis set. NMR spectra for both enantiomers of each epoxide product were simulated and found to be the same. Hence, separate spectral assignments were not necessary.
All spectra were found to match closely with the literature.[9]
Styrene Oxide
NMR spectra: DOI:10042/25777 NMR couplings: DOI:10042/25778
1H NMR
-epoxide_1-H_NMR.svg)
-styrene_oxide_1-H_NMR.jpg)
δ 2.54 (1H, dd, 2J(Ha-Hb) 5.6 Hz, 3J(Ha-Hc) 1.9 Hz, a), 3.12 (1H, dd, 2J(Ha-Hb) 5.6 Hz, 3J(Hb-Hc) 4.4 Hz, b), 3.66 (1H, dd, 3J(Ha-Hc) 1.9 Hz, 3J(Hb-Hc) 4.4 Hz, c), 7.30-7.49 (5H, m, d-h)
13C NMR
-epoxide_13-C_NMR.svg)
-styrene_oxide_13-C_NMR.jpg)
δ 53.5 (1), 54.1 (2), 118.3 (8), 123.0 (4, 6), 123.4 (5), 124.1 (7), 135.1 (3)
trans-β-Methylstyrene Oxide
NMR spectra: DOI:10042/25779 NMR couplings: DOI:10042/25780
1H NMR
![]()
![]()
δ 1.33 (3H, d, 2J(Ha-Hb) 5.4 Hz, b), 2.79 (1H, dq, 2J(Ha-Hb) 5.4 Hz, 3J(Ha-Hc) 1.4 Hz, a), 3.41 (1H, d, 3J(Ha-Hc) 1.4 Hz, c), 7.31-7.49 (5H, m, d-h)
13C NMR
![]()
![]()
δ 18.8 (1), 60.6 (3), 62.3 (2), 118.4 (9), 122.7 (7), 122.8 (5), 123.3 (6), 124.1 (8), 135.0 (4)
Assigning the Absolute Configuration of the Epoxide Products
Optical Rotation
(R)-Styrene OR: DOI:10042/25781 (S)-Styrene OR: DOI:10042/25782 (R),(R)-tran-β-methylstyrene OR: DOI:10042/25783 (S),(S)-tran-β-methylstyrene OR: DOI:10042/25784
The outputs from the NMR calculations containing the optimised structures were used to predict the optical rotation (OR) for each of the epoxide enantionmers. The B3LYP density functional method was used, with chloroform as the solvent. Comparison with the literature[10] has been made below, however all OR values are below 100 deg and hence cannot be used to predict absolute configurations.
| Styrene / deg | trans-β-methylstyrene / deg |
|---|---|
| (R) -30.44 | (R),(R) +46.77 |
| (S) +30.43 (lit. +32.1) | (S),(S) -46.77 (lit. -41.8) |
Note: literature conditions - 1 M solutions, CHCl3, 589nm light, 25 deg C, 99% ee
Using the Calculated Properties of the Transition State
The asymmetric epoxidation reaction takes place under kinetic control and the major product will be formed via the lowest energy transition state. Therefore, analysis of the transition state energies allows one to predict the major enantiomeric product and the enantiomeric excess of the reaction. In this discussion, the transition states for the Shi epoxidation of trans-β-methylstyrene and Jacobsen epoxidation of cis-β-methylstyrene are analysed based on quantum mechanical calculations of the energy. The total energy of each transition state system was corrected for entropy and zero-point thermal energies and was simulated in aqueous conditions.
Shi Epoxidation of trans-β-methylstyrene
For the Shi epoxidation of trans-β-methylstyrene, there are eight possible transition states. The specific transition state geometry depends on whether the (R),(R) or (S),(S)-epoxide is being formed, which of the two dioxirane oxygens is transferred (O1 or O2 labelled below) and whether the phenyl group is positioned endo or exo relative to the catalyst molecule. The table below shows the energies of each of the transition states relative to the energy of the lowest energy transition state: (R),(R)-epoxide, O1 transfer, endo phenyl.

| Transition State | Relative Total Energy / kJ/mol | |
|---|---|---|
| (R),(R)-epoxide | (S),(S)-epoxide | |
| O2 transfer, endo Ph | 24.871 | 44.213 |
| O2 transfer, exo Ph | 34.683 | 38.072 |
| O1 transfer, exo Ph | 8.325 | 20.219 |
| O1 transfer, endo Ph | 0.000 | 22.781 |
The lowest energy transition state occurs in the formation of the (R),(R)-epoxide, and hence is in agreement with the experimental findings of Wong et al.,[3] which indicate this is the major product. This transition state is particularly stable because the alkene substituents are far away in space from the ketal groups on the catalyst and hence do not suffer from steric repulsion. In addition, it is clear that the transfer of oxygen 1 leads to lower energy transition states than found for the transfer of oxygen 2. In all cases, the transfer of oxygen 2 forces close contact between the dimethyl ketal group in ring A of the catalyst and one of the alkene substituents, leading to repulsive interactions in the transition state. Conversely, the orientation of the phenyl group (endo or exo) leads to both bonding and repulsive interactions in the transition states, depending on whether the position of the phenyl group results in favourable van der Waals contacts or unfavourable steric repulsions.
Predicting the Enantiomeric Excess (ee) of the Shi epoxidation
The ee for the Shi epoxidation of trans-β-methylstyrene will be predicted by comparing the energy difference between the lowest energy (R),(R) pathway and the lowest energy (S),(S) pathway. This energy difference can then be converted into a ratio of concentrations (K) of the two species and hence allowing the ee to be calculated.
and therefore
Hence
This calculated ee is similar to the experimental ee of 95.5% observed by Wang et al.[4] The theoretical ee is expected to be greater than the experimental ee as the Shi catalyst used for the reaction is unlikely to be 100% pure.
Jacobsen Epoxidation of cis-β-methylstyrene
For the Jacobsen epoxidation of cis-β-methylstyrene, there are four possible transition states that depend on whether the (S),(R) or (R),(S)-epoxide is formed and whether the phenyl is endo or exo relative to the catalyst. Again, the table below shows the energies of each transition state relative to the energy of the lowest energy transition state: (S,R)-epoxide, endo phenyl.
| Transition State | Relative Total Energy / kJ/mol | |
|---|---|---|
| (S),(R)-epoxide | (R),(S)-epoxide | |
| endo Ph | 0.000 | 22.314 |
| exo Ph | 16.060 | 24.388 |
The lowest energy transition state occurs for the (S),(R)-epoxide and hence this product is formed in enantiomeric excess. This is in agreement with the literature.[7] In addition, the endo geometry was found to result in a more stable transition state for both epoxide products. The endo orientation positions the alkene phenyl group close to the aromatic rings on catalyst and presumably leads to the formation of favourable van der Waals contacts.
Predicting the Enantiomeric Excess (ee) of the Jacobsen epoxidation
Again, the energy difference between the lowest energy (S),(R) pathway is compared with the lowest energy (R),(S) pathway in order to calculate the ee of the epoxidation reaction.
and therefore
Hence
The predicted ee is higher for the Jacobsen catalyst when compared to the Shi catalyst, stemming from the larger energy gap present within in the Jacobsen system. The observed ee in the literature by Jacobsen et al. was 92%, and hence the calculated ee is again relatively similar.
Investigating the Non-covalent Interactions in the Active Site of the Reaction Transition State
The most stable transition state for the Shi epoxidation of trans-β-methylstyrene ((R),(R)-epoxide, O1 transfer, endo phenyl) was subjected to NCI analysis. The two diagrams below are different perspectives of the same transition state (left = side-on view; right = top-down view). The colours indicate whether the interaction is attractive (blue), moderately attractive (green), moderately repulsive (yellow) or strongly repulsive (red).


Interaction 1 is a strongly attractive van der Waals contact between the substrate's phenyl ring and an axial hydrogen on the central six-membered ring of the catalyst. Interactions 2 and 3 indicate there are moderately attractive dipolar interactions between two olefinic hydrogens on the substrate and the oxygens atoms in one of the five-membered rings of the catalyst. The methyl hydrogens on the substrate also contribute to the observed interaction at position 3. Finally, interaction 4 is the bond forming region of the transition state and hence is not considered an NCI. Overall, it can be concluded that the attractive van der Waals contacts between the endo phenyl group and the catalyst molecule are the primary reason why a particularly low energy transition state is observed for this system.
Investigating the Electronic Topology (QTAIM) in the Active Site of the Reaction Transition State
The same transition state as analysed above for NCIs ((R),(R)-epoxide, O1 transfer, endo phenyl) was subjected to QTAIM analysis. The diagram below shows three interesting regions containing bond critical points (BCPs).

Arrows 1 and 2 point towards non-covalent interactions. Interaction 1 is an attractive van der Waals contact between the substrate and catalyst, with the BCP being placed slightly closer to the axial hydrogen than the phenyl carbon. The interactions indicated by arrow 2 are weak interactions between an oxygen atom on the catalyst and phenyl and alkene hydrogen atoms on the substrate. Here, the BCPs lie approximately half way between the two atoms. The interactions indicated by 3 are associated with covalent bonds. The atom in the centre of the box is the anomeric carbon, which is directly bound to two oxygen atoms. The BCPs lie much closer to the carbon atom than the oxygen atoms in the C-O bonds, indicating that these covalent bonds are polarised.
Suggesting a new candidate for investigation

(1R,4S)-pulegone oxide has an optical rotatory power of 562.3 deg according to the literature.[11] In addition, the alkene from which is it derived is commercially available, and hence the epoxide is a suitable candidate for investigation. The ketone may be reduced to an alcohol to form the allylic alcohol, providing the opportunity to investigate other catalytic systems such as the Sharpless asymmetric epoxidation.
References
- ↑ Y. Tu, Z. Wang and Y. Shi, J. Am. Chem. Soc., 1996, 118 (40), 9806-9807. DOI:10.1021/ja962345g
- ↑ M. Ďurík et al., Acta Cryst., 2001, 672-674. DOI:10.1107/S160053680101073X
- ↑ 3.0 3.1 O. A. Wong, B. Wang, M. Zhao and Y. Shi, J. Org. Chem., 2009, 74 (16), 6335-6338. DOI:10.1021/jo900739q
- ↑ 4.0 4.1 Z. Wang, Y. Tu, M. Frohn, J. Zhang and Y. Shi, J. Am. Chem. Soc., 2009, 119 (46), 11224-11235. DOI:10.1021/ja972272g
- ↑ M. Palucki et al., J. Am. Chem. Soc., 1998, 120 (5), 948-954. DOI:10.1021/ja973468j
- ↑ J. W. Yoon, T. S. Yoon, S. W. Lee and W. Shin, Acta Cryst., 1999, 1766-1769. DOI:10.1107/S0108270199009397
- ↑ 7.0 7.1 E. N. Jacobsen, W. Zhang, A. R. Muci, J. R. Ecker and L. Deng, J. Am. Chem. Soc., 1991, 113 (18), 7063-7064. DOI:10.1021/ja00018a068
- ↑ B. D. Brandes and E. N. Jacobsen, J. Org. Chem., 1994, 59 (16), 4378-4380. DOI:10.1021/jo00095a009
- ↑ L. Ji, Y. Wang, C. Qian and X. Chen, Synth. Comm., 2013, 43 (16), 2256-2264. DOI:10.1080/00397911.2012.699578
- ↑ H. Lin, Y. Liu and Z. Wu, Tetrahedron: Asymmetry, 2011, 22 (2), 134-137. DOI:10.1016/j.tetasy.2010.12.022
- ↑ W. Reusch and C. K. Johnson, J. Org. Chem., 1963, 28 (10), 2557–2560. DOI:10.1021/jo01045a016