
Rep:Mod:Td81121mini
Introduction
Ionic liquids are an interesting class of materials due to their ability to remain in the liquid state at relatively low temperatures (i.e. <100o) despite being a salt. They also offer important industrial applications such as in the BASIL (Biphasic Acid Scavenging utilising Ionic Liquids) process by BASF, where '1-alkylimidazon scavenged acid from existing processes', resulting in formation of ionic liquids which can be easily removed hence enhancing reaction rates, increasing yields and improving selectivity[1].
Due to the vast amount of possible combinations of compounds that could be used to make ionic liquids, computational chemistry is increasingly becoming an important tool for investigations. In this Inorganic Computational Mini Project, titled "Inorganic Liquids: Designer Solvents", an investigation into possible candidates as the cation component for ionic liquids was carried out. Computational calculations, including optimisation, frequency analysis, MO and NBO analysis, were conducted initially for three different 'onium' cations ([N(CH3)4]+, [P(CH3)4]+, [S(CH3)3]+) in order to compare and contract them. Thereafter, influences of functional groups were studied by carrying out calculations on [N(CH3)(CH2OH)]+ and [N(CH3)(CH2CN)]+.
Part 1: Comparison of selected 'onium' cations
Optimisation, frequency analysis, MO and NBO calculations were carried out using Gaussian on three 'onium' ions, [N(CH3)4]+, [P(CH3)4]+ and [S(CH3)3]+. The information from these calculations were then used to compare the structures.
Optimisation
The three onium cations were optimised using the DFT method with 6-31G basis set (with charge=1) and the keywords: "opt=tight, int=ultrafine, scf-conver=9". In addition, the "nosymm" keyword was used in order to prevent the geometry being constrained and hence obtaining a better set of calculations. The results are listed below.
| Summary Table | |||
|---|---|---|---|
| Molecule | 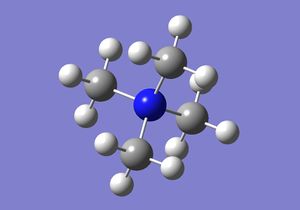 |
4_image.jpg) |
3_image.jpg) |
| File Name | TC_N(CH3)4_opt_631g_d_p | TC_P(CH3)4_opt_631g_d_p | TC_S(CH3)3_opt_631g_d_p |
| File Type | .log | .log | .log |
| Calculation Type | FOPT | FOPT | FOPT |
| Calculation Method | RB3LYP | RB3LYP | RB3LYP |
| Basis Set | 6-31G(d,p) | 6-31G(d,p) | 6-31G(d,p) |
| Charge | 1 | 1 | 1 |
| Spin | Singlet | Singlet | Singlet |
| Energy (a.u.) | -214.18127322 | -500.82701172 | -517.68327361 |
| RMS Gradient Norm | 0.00000003 | 0.00000108 | 0.00000135 |
| Dipole Moment (D) | 14.26 | 14.26 | 5.89 |
| Point Group | C1 | C1 | C1 |
| Calculation Time | 12 min 22.3 Sec | 24 min 2.2 sec | 9 min |
| C-X Bond Distance (Å) | 1.51 | 1.82 | 1.82 |
| C-X-C Bond Angle | 109.5o | 109.5o | 102.7o |
| Log File | [[N(CH3)4+]] | [[P(CH3)4+]] | [[S(CH3)3+]] |
From the calculation data, it can be seen that [N(CH3)4]+ has a C-N bond length of 1.51 Å. This is close to the published experimental value of 1.49Å [2]. Possible improvements could be made by using a larger basis set. In addition, the C-N-C bond angle was determined to be 109.5o, in accordance with the angle for a tetrahedral molecule.
The C-P bond distance was found to be 1.82Å from the calculations. The literature value for a related compound P(CH3)3 was found to be 1.85Å [3]. The two values are again fairly close. The C-P-C bond angle was determined to be the expected 109.5o, that of a tetrahedral molecule.
The computational study yielded a C-S bond distance of 1.82Å. This is identical to the literature value [4]. However, the C-P-C bond angle was shown to be 102.7o, about four degrees less than that of the expected 107o for a trigonal pyramidal molecule. Normally, one might expect the angle to be slightly larger than the typical trigonal pyramidal geometry angle (c.f. ~106o C-N-C bond angle of NH3 - results data in week one compulsory part wiki) as sulphur is a bigger atom and would therefore have more space around it. However, the opposite is observed. A possible explanation for this could be that because sulphur is a larger atom, it has more diffused orbitals and because it has a lower electronegativity value compared to nitrogen, it does not draw as much electron density towards itself. The result is that there appear to be more electron density at the opposite side of the three methyl groups in [S(CH3)3]+. Because of electron repulsion between the lone pair and the bonding pairs, effectively the three methyl groups are pushed closer together, hence the molecule has a smaller than expected bond angle.
| Molecule | Item Table |
|---|---|
| [N(CH3)4]+ | Item Value Threshold Converged?
Maximum Force 0.000000 0.000015 YES
RMS Force 0.000000 0.000010 YES
Maximum Displacement 0.000008 0.000060 YES
RMS Displacement 0.000003 0.000040 YES
Predicted change in Energy=-5.949320D-13
Optimization completed.
-- Stationary point found.
|
| [P(CH3)4]+] | Item Value Threshold Converged?
Maximum Force 0.000003 0.000015 YES
RMS Force 0.000000 0.000010 YES
Maximum Displacement 0.000054 0.000060 YES
RMS Displacement 0.000015 0.000040 YES
Predicted change in Energy=-5.512326D-11
Optimization completed.
-- Stationary point found.
|
| [S(CH3)3]+ | Item Value Threshold Converged?
Maximum Force 0.000002 0.000015 YES
RMS Force 0.000001 0.000010 YES
Maximum Displacement 0.000030 0.000060 YES
RMS Displacement 0.000012 0.000040 YES
Predicted change in Energy=-1.163089D-10
Optimization completed.
-- Stationary point found.
|
Frequency Analysis
The frequency analysis was carried out with the optimised molecule above with the basis set 6-31G(d,p) and keywords: "opt=tight, int=ultrafine, scf=conver=9 and nosymm". The low frequency results are well within the required boundaries of ±20-30cm-1 and no negative imaginary vibrational frequencies were observed, thereby indicating the calculations have indeed obtained appropriate minima for the three cations.
| molecule | Low Frequency | IR Spectrum | Log File |
|---|---|---|---|
| [N(CH3)4]+ | Low frequencies --- -5.4462 -2.0704 -0.0009 0.0004 0.0008 3.9335 Low frequencies --- 183.7602 288.3981 288.8993 |
4_IR_spectrum.jpeg) |
[[N(CH3)4+]] |
| [P(CH3)4]+ | Low frequencies --- -2.5047 -0.0037 -0.0023 0.0009 5.1165 7.5702 Low frequencies --- 156.4500 192.0500 192.2801 |
4_IR_spectrum.jpeg) |
[[P(CH3)4+]] |
| [S(CH3)3]+ | Low frequencies --- -9.4807 -3.6576 0.0019 0.0026 0.0044 3.6442 Low frequencies --- 162.0547 199.5452 199.7009 |
3_IR_spectrum.jpeg) |
[[P(CH3)3+]] |
MO Analysis
Molecular Orbital analysis was carried out on the three cations using the previously optimised structures. The population analysis can be found in the table below:
| Molecule | Population Analysis .log file |
|---|---|
| N(CH3)4]+ | [N(CH3)4+ log file]] |
| P(CH3)4]+ | [P(CH3)4+ log file]] |
| S(CH3)3]+ | [S(CH3)3+ log file]] |
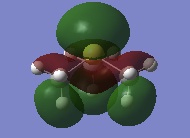
Below is a selection of five occupied non-core MOs (determined from the amount of energy associated and specific structure) from the [N(CH3)4]+ molecule ranging from highly bonding to highly antibonding.
| MO No. | MO depiction | Description | Possible LCAO MOs |
|---|---|---|---|
| 6 | 4_MO_6_annotated.jpg) |
Strong bonding interactions exists between all of the atoms in the lowest non-core MO, possibly due to in-phase overlap between the S orbitals. In addition, weak through space bonding interactions exist making this MO a higher bonding MO. The calculation suggests a very delocalised MO, as the electron density appear to be spread over almost all of the atoms. | 4_MO_6_LCAO.jpg) |
| 10 | 4_MO_10_annotated.jpg) |
Even though there is antibonding interaction between the central nitrogen atom and the four carbons, thereby forming four nodes, bonding AO interactions exist between all of the methyl ligands. In addition, there is also through space bonding interactions between adjacent CH3 groups. Overall, the MO is likely to be bonding or weakly bonding. | 4_MO_10_LCAO.jpg) |
| 13 | 4_MO_13_annotated.jpg) |
Bonding interaction exists between the pz orbitals of the central nitrogen and carbons. In addition, further bonding interactions exists between the p orbital of the carbon and the s orbitals of the bonded hydrogens. Nevertheless, two nodal planes does exist with several through space weakly antibonding interactions, making this a weakly bonding MO. | 4_MO_13_LCAO.jpg) |
| 16 | 4_MO_16_annotated.jpg) |
Four nodes exist in this antibonding MO. Although there are a bonding interactions between the p orbitals of the carbon and s orbitals of the hydrogen, these interactions are fairly weak, as characterised by the small overlap. Furthermore, there are many through space antibonding interactions between neighbouring methyl groups. Overall, it is likely to be an antibonding MO. | 4_MO_16_LCAO.jpg) |
| 21 | 4_MO_21_annotated_.jpg) |
p orbital in-phase overlap exists between the carbons and nitrogen. In addition to bonding overall between the carbons and their hydrogens, it appears that a significant amount of mixing exists between the axial hydrogens and relatively smaller amount of mixing exists between the equatorial hydrogens. The result is a fairly delocalised MO. Three nodal planes exists in the MO, together with significant amount of through space antibonding interactions makes this MO weakly bonding. | 4_MO_21_LCAO.jpg) |
NBO Analysis
The NBO study was performed using the .log file from the population analysis calculations. The results are tabulated below (note: all charge range are from -0.500 to 0.500).
| NBO analysis of [N(CH3)4]+] | |
|---|---|
4_NBO_image.jpg)
| |
| Atom | Charge |
| N | -0.295 |
| C | -0.483 |
| H | +0.269 |
Contrary to the traditional picture of [NR4]+, the analysis indicates that the positive charge resides on the hydrogen atoms and the nitrogen actually bears a negative charge. This is due to the fact that, although the nitrogen donated its lone pair to form the extra C-N bond, because it is highly electronegative, it attracts most of the electron density from the bond it forms, thereby resulting in a negatively charged nitrogen. The carbons are also negatively charged as it also pull the majority of the electron density from the C-H bonds towards itself as it is more electronegative than hydrogen. The hydrogen, therefore, ends up positively charged. The overall charge of the molecule sums up to +1, as expected.
| NBO analysis of [P(CH3)4]+] | |
|---|---|
4_NBO_image.jpg)
| |
| Atom | Charge |
| P | +1.667 |
| C | -1.060 |
| H | +0.298 |
In the molecule [P(CH3]+, phosphorus is less electronegative than carbon. This results in phosphorus being relatively higher positively charged and the carbon bearing the majority of the electron density, as it also draws additional electron density from its substituent hydrogens. Again because of the electron pull from the carbons via inductive effects, the hydrogens bear a positive charge. The total charge of the molecule is +1.
| NBO analysis of [S(CH3)3]+] | |
|---|---|
3_NBO_image.jpg)
| |
| Atom | Charge |
| S | +0.917 |
| C | -0.846 |
| H (4,8,12) | +0.279 |
| H (1,2,3,5,6,7,9,10,11) | +0.297 |
The carbon atoms in the [S(CH3]3]+ bears all of the negative charge as it draws most of the electron density from its neighbouring hydrogens through inductive effects. The sulphur has a smaller electronegativity value compared to nitrogen, hence it is better at donating its lone pair. As such, the central sulphur bears a positive charge. A feature that makes this molecule different from the two before it is that, not all the hydrogens carry the same amount of positive charge. In particular, the hydrogens numbered 4,8 and 12 (axial hydrogens) have a slightly lower positive charge than the rest of the other hydrogens. A possible reason could be that the axial hydrogens are arranged so that the σ* C-H bond is anti-periplanar (app) to the lone pair of the sulphur. This app arrangement facilitates stereoelectronc effects, in that the sulphur orbital that is properly aligned donates electron density into the C-H bond. The result can be seen as the C-H(4,8,12) bond length are actually different than the other C-H bonds. In addition, the Second Order Perturbation Theory Analysis of Fock Matrix in the NBO analysis also highlights the fact that the bond interaction energy for the axial-hydrogen to carbon bonds are 2.31 kcal/mol. These factors result in the axial hydrogens having slightly higher electron density compared to the other hydrogens and hence a lower positive charge.
| Hydrogen type/number | C-H Bond length (Å) |
|---|---|
| Axial / H(4,8,12) | 1.09139 |
| Equatorial / H(1,2,3,5,6,7,9,10,11) | 1.09158 |
Please note: gaussian may have interpreted the bonds to be shorter due to more electron density in the bond resulting from donation from the sulphur lone pair, but in actual fact, it is donation into the σ* C-H bond and should therefore, potentially, weaken the bond. Even though keywords have been used in the calculations, the basis set is probably not good enough. As such, a higher basis set may be able to correct for this error as the difference between the bond lengths is very small. In order words, the information on bond length, because it is quoted to such high decimal places should not be taken to be accurate, but rather, the fact that there are slight differences in bond length should be the main focus.
Second Order Perturbation Theory Analysis of Fock Matrix in NBO Basis
Donor NBO (i) Acceptor NBO (j) kcal/mol a.u. a.u. ================================================================================================= 21. LP ( 1) S 13 /100. BD*( 1) C 1 - H 4 2.32 1.02 0.043 21. LP ( 1) S 13 /104. BD*( 1) C 5 - H 8 2.32 1.02 0.043 21. LP ( 1) S 13 /108. BD*( 1) C 9 - H 12 2.32 1.02 0.043
Orbital Contribution to C-X Bond
The population analysis was also used to provide additional information to investigate the relative contributions of the C and heteroatom to the C-X Bond. In particular, the results from the "(Occupancy) Bond orbital/ Coefficients/ Hybrids table" was employed to explore the electron contributions from each atom. The results are listed below.
| molecule | Orbital Contribution Result |
|---|---|
| [N(CH3)4]+ |
(Occupancy) Bond orbital/ Coefficients/ Hybrids
---------------------------------------------------------------------------------
4. (1.98452) BD ( 1) C 1 - N 17
( 33.65%) 0.5801* C 1 s( 20.78%)p 3.81( 79.06%)d 0.01( 0.16%)
-0.0003 -0.4552 0.0237 -0.0026 -0.8884
-0.0377 -0.0001 0.0000 -0.0001 0.0000
0.0000 0.0000 0.0000 -0.0352 0.0203
( 66.35%) 0.8146* N 17 s( 25.00%)p 3.00( 74.97%)d 0.00( 0.03%)
0.0000 -0.5000 0.0007 0.0000 0.8658
-0.0001 0.0001 0.0000 0.0001 0.0000
0.0000 0.0000 0.0000 -0.0154 0.0089
|
| [P(CH3)4]+ |
(Occupancy) Bond orbital/ Coefficients/ Hybrids
---------------------------------------------------------------------------------
4. (1.98030) BD ( 1) C 1 - P 17
( 59.57%) 0.7718* C 1 s( 25.24%)p 2.96( 74.67%)d 0.00( 0.08%)
0.0002 0.5021 0.0171 -0.0020 0.8640
-0.0158 -0.0001 0.0000 0.0001 0.0000
0.0000 0.0000 0.0000 0.0251 -0.0145
( 40.43%) 0.6358* P 17 s( 25.00%)p 2.97( 74.15%)d 0.03( 0.85%)
0.0000 0.0001 0.5000 -0.0008 0.0000
0.0000 -0.8611 0.0012 0.0000 0.0000
0.0000 0.0000 -0.0001 0.0000 0.0000
0.0000 0.0000 0.0800 -0.0462
|
| [S(CH3)3]+ |
(Occupancy) Bond orbital/ Coefficients/ Hybrids
---------------------------------------------------------------------------------
4. (1.98631) BD ( 1) C 1 - S 13
( 48.67%) 0.6976* C 1 s( 19.71%)p 4.07( 80.16%)d 0.01( 0.14%)
0.0003 0.4437 0.0140 -0.0033 -0.3635
-0.0098 -0.4089 0.0032 0.7085 -0.0055
0.0120 -0.0208 -0.0230 -0.0017 0.0163
( 51.33%) 0.7164* S 13 s( 16.95%)p 4.86( 82.42%)d 0.04( 0.63%)
0.0000 0.0001 0.4117 -0.0076 0.0012
0.0000 0.4039 0.0259 0.0000 0.4057
-0.0179 0.0000 -0.7032 0.0309 0.0310
-0.0537 -0.0428 0.0080 0.0240
|
From the data, it can be seen that for the [N(CH3)4]+ molecule, most of the electron density comes from the nitrogen atom (66.35%). The NBO analysis outlines the electron density of the entire molecule by its atomic like orbitals. This information is then used to from 2e-2c bonds. In the traditional picture of [NR4]+, the nitrogen bears a positive charge, as it indicates formation of a covalent bond via donation of nitrogen's lone pair and that all atoms deliver electron equally. However, as the computational study has suggested, nitrogen actually has most of the electron density. As such, it ends up carrying a negative charge, which is also in accordance with the analysis performed for the charge distribution.
For the [P(CH3)4]+ molecule, the carbon atom delivers most of the electron density, as from the analysis, phosphorus' orbital contribution to the C-P bond is only 40.43%. Furthermore, due to the difference in electronegativity, as explained in the charge distribution analysis above, this results in phosphorous bearing a highly positive charge.
In the [S(CH3]+ molecule, because the electronegativity is similar between sulphur and carbon, the orbital contribution to the C-S bond is roughly equal, with sulphur delivering 51.33%. This results in roughly a +1 charge on sulphur.
Part 2: Influence of functional groups
In this part of the investigation, two additional molecules, [N(CH3)3(CH2OH]+ and [N(CH3)3(CH2CN)]+, have been optimised and additional frequency analysis, MO and NBO calculations were carried out on them. The result, mainly the HOMO and the LUMO, was used to compare the structures as well as that of [N(CH3)4]+ from part 1.
Optimisation
The optimisation process was performed on [N(CH3)3(CH2OH)]+ and [N(CH3)3(CH2CN)]+ using the basis set 6-31G(d,p) and method B3LYP. For the former molecule, the dihedral angle between the nitrogen and the hydrogen of the -OH group, looking down the connecting C-O bond had to be distorted in order to obtain the true optimised structure, as proven by the frequency analysis (more information in supplementary information section below).
| Summary Table | ||
|---|---|---|
| Molecule | (CH2OH)_image.jpg) |
3(CH2CN)_image.jpg) |
| File Name | TC_N(CH3)3(CH2OH)_opt_631g_d_p_symm_distort | TC_N(CH3)3(CH2CN)_opt_631g_d_p |
| File Type | .log | .log |
| Calculation Type | FOPT | FOPT |
| Calculation Method | RB3LYP | RB3LYP |
| Basis Set | 6-31G(d,p) | 6-31G(d,p) |
| Charge | 1 | 1 |
| Spin | Singlet | Singlet |
| Energy | -289.39470639 a.u. | -306.39375974 a.u. |
| RMS Gradient Norm | 0.00000072 | 0.00000039 |
| Dipole Moment | 14.41 D | 14.60 D |
| Point Group | C1 | C1 |
| Calculation Time | 46 min 27.8 sec | 24 min 6.5 sec |
| log file | [[N(CH3)3(CH2OH)]+] | [[N(CH3)3(CH2CN)]+] |
| Molecule | Item Table |
|---|---|
| [N(CH3)3(CH2OH)]+] |
Item Value Threshold Converged?
Maximum Force 0.000001 0.000015 YES
RMS Force 0.000000 0.000010 YES
Maximum Displacement 0.000025 0.000060 YES
RMS Displacement 0.000006 0.000040 YES
Predicted change in Energy=-1.456031D-11
Optimization completed.
-- Stationary point found.
|
| [P(CH3)4]+] |
Item Value Threshold Converged?
Maximum Force 0.000001 0.000015 YES
RMS Force 0.000000 0.000010 YES
Maximum Displacement 0.000026 0.000060 YES
RMS Displacement 0.000007 0.000040 YES
Predicted change in Energy=-1.708158D-11
Optimization completed.
-- Stationary point found.
|
Frequency Analysis
The frequency analysis was carried out with the optimised molecules above and the basis set 6-31G(d,p), method B3LYP with keywords: "opt=tight, int=ultrafine, scf=conver=9 and nosymm". The low frequencies obtained were all within the appropriate limit and no imaginary frequencies were obtained ([N(CH3)3(CH2OH)]]+ molecule required slight change in the dihedral angle between the -OH and the N atom, please see supplementary section for more information).
| molecule | Low Frequency | IR Spectrum | Log File |
|---|---|---|---|
| [N(CH3)3(CH2OH]+ | Low frequencies --- -11.1623 -3.3470 -2.7452 -0.0011 0.0010 0.0011 Low frequencies --- 131.0191 213.9810 255.7910 |
3(CH2OH)_IR_spectrum.jpeg) |
[[N(CH3)3(CH2OH)]+] |
| [N(CH3)3(CH2CN]+ | Low frequencies --- -8.2127 -6.4955 -3.6484 -0.0002 0.0003 0.0005 Low frequencies --- 91.7524 153.7797 211.1726 |
3(CH2CN)_IR_spectrum.jpeg) |
[[N(CH3)3(CH2CN)]+] |
MO
The population analysis was performed using the above optimised molecules [N(CH3)3(CH2OH)]+ and [N(CH3)3(CH2CN)]+ with the same basis set and method with the addition keyword: "pop=full". The .log files of these analysis can be seen below:
| Molecule | .log file |
|---|---|
| [N(CH3)3(CH2OH)]+ | [[N(CH3)3(CH2OH)+ .log file]] |
| [N(CH3)3(CH2CN)]+ | [[N(CH3)3(CH2CN)+ .log file]] |
NBO
The NBO analysis was performed using the population calculation .log file. The charge range was manually set to -0.5 to 0.5 for both molecules. The charge distribution is listed below.
| [N(CH3)3(CH2OH)]+ Charge Distribution | [N(CH3)3(CH2OH)]+ Atom Label diagram | label number | Atom | Charge |
|---|---|---|---|---|
3(CH2OH)_NBO_image.jpg) |
3(CH2OH)_atom_label_image.jpg) |
1 | C | -0.494 |
| 2 | H | +0.272 | ||
| 3 | H | +0.271 | ||
| 4 | H | +0.262 | ||
| 5 | C | +0.088 | ||
| 6 | H | +0.249 | ||
| 7 | H | +0.237 | ||
| 8 | C | -0.492 | ||
| 9 | H | +0.269 | ||
| 10 | H | +0.274 | ||
| 11 | H | 0.266 | ||
| 12 | C | -0.491 | ||
| 13 | H | +0.266 | ||
| 14 | H | 0.282 | ||
| 15 | H | 0.266 | ||
| 16 | N | -0.322 | ||
| 17 | O | -0.725 | ||
| 18 | H | 0.521 |
(CH2OH)_MO_7_NCO_delocalisation_.jpg)
The substitution of an -OH group for a hydrogen of a methyl group has a pronounced effect on the charge distribution of the molecule. The hydroxyl group is electron donating, as a result the central nitrogen now bears a higher negative charge compared to the original [N(CH3)4]3 molecule. This is, most likely, due to a high degree of electron density delocalisation between the nitrogen, carbon (number 5) and oxygen, which is facilitated by the app arrangement between the oxygen lone pair and the C-N bond.
Perhaps the most pronounced effect is the change of charge distribution of the carbon bearing the new -OH group. The carbon (number 5) has changed from formerly negatively charged in the [N(CH3)4]3 molecule to a slightly positively charged carbon. This is due to the oxygen being highly eletronegative and hence drawing electron density way from the carbon via inductive effects.
The charge distribution of the hydrogens (atom label 6 & 7) connected to the carbon bearing the -OH group has also changed. It can be observed that the positive charge on these hydrogens have now diminished slightly. In particular, the effect can be seen to be greater for one of the hydrogens (hydrogen number 7). The reason is likely to do with donation of electron density from the oxygen lone pair to the hydrogens in question. The reason for the effect to be slightly greater for hydrogen number 7 is likely to be because there is better overlap between the oxygen's lone pair orbital and the hydrogen number 7's orbital, hence resulting in a slightly lower positive charge compared to hydrogen number 6.
| Second Order Perturbation Theory Analysis of Fock Matrix in NBO Basis data | Comments |
|---|---|
Donor NBO (i) Acceptor NBO (j) kcal/mol a.u. a.u.
===================================================================================================
25. LP ( 2) O 17 /140. BD*( 1) C 5 - N 16 18.96 0.51 0.088
|
Strong interaction between the oxygen lone pair and the C-N bond; indication of a strong delocalisation of the lone pair into the O-C-N fragment of the molecule |
Donor NBO (i) Acceptor NBO (j) kcal/mol a.u. a.u. =================================================================================================== 24. LP ( 1) O 17 /138. BD*( 1) C 5 - H 6 1.21 1.04 0.032 24. LP ( 1) O 17 /139. BD*( 1) C 5 - H 7 3.97 1.02 0.057 25. LP ( 2) O 17 /138. BD*( 1) C 5 - H 6 2.97 0.77 0.043 25. LP ( 2) O 17 /139. BD*( 1) C 5 - H 7 0.83 0.75 0.023 |
Lone pair donation from the oxygen into the hydrogen number 6 & 7 can be observed from the data. The effect is slightly greater for hydrogen number 7 |
| [N(CH3)3(CH2CN)]+ Charge Distribution | [N(CH3)3(CH2OH)]+ Atom Label diagram | label number | Atom | Charge |
|---|---|---|---|---|
3(CH2CN)_NBO_image.jpg) |
3(CH2CN)_atom_label_image.jpg) |
1 | C | -0.489 |
| 2 | H | +0.274 | ||
| 3 | H | +0.282 | ||
| 4 | H | +0.269 | ||
| 5 | C | -0.358 | ||
| 6 | H | +0.309 | ||
| 7 | H | +0.309 | ||
| 8 | C | -0.485 | ||
| 9 | H | +0.271 | ||
| 10 | H | +0.277 | ||
| 11 | H | 0.271 | ||
| 12 | C | -0.489 | ||
| 13 | H | +0.269 | ||
| 14 | H | 0.282 | ||
| 15 | H | 0.274 | ||
| 16 | N | -0.289 | ||
| 17 | C | -0.209 | ||
| 18 | N | -0.186 |
The addition of a -CN, electron withdrawing group, has changed the charge distribution compared to the original [N(CH3)4]+. In terms of the constituent atoms that make up the -CN group, the nitrogen bears the majority of the electron density, due to its higher eletronegatively, resulting in the nitrogen and carbon atom carrying a negative and positive charge respectively. Although the central nitrogen atom is not affect by the -CN group, the CH2 neighbouring the nitrile group is massively affected compared to the other methyl groups. The carbon bears a slightly lower negative charge, due to the electron withdrawing effects of the -CN group. The hydrogens (atom label 6 & 7) are slightly more positive than the hydrogens in the methyl groups. This is, again, due to the nitrile group pulling electron density aways from the hydrogens.
Comparison of HOMO and LUMO of [N(CH3)4]+ [N(CH3)3(CH2OH)]+ and [N(CH3)3CH2CN]+
A comparison between the HOMO and LUMO of [N(CH3)4]+ [N(CH3)3(CH2OH)]+ and [N(CH3)3CH2CN]+ are outlined below.
| Molecule | [N(CH3)4]+ | [N(CH3)3(CH2OH)]+ | [N(CH3)3CH2CN]+ | |||
|---|---|---|---|---|---|---|
| Orbital Type | HOMO | LUMO | HOMO | LUMO | HOMO | LUMO |
| Visualisation | 4_HOMO_image.jpg) |
4_LUMO_image.jpg) |
3(CH2OH)_HOMO_image.jpg) |
(CH2OH)_LUMO_image.jpg) |
3(CH3CN)_HOMO_image.jpg) |
3(CH2CN)_LUMO_image.jpg)
|
| Energy (a.u.) | -0.57934 | -0.13302 | -0.48763 | -0.12459 | -0.50047 | -0.18182 |
The introduction of functional groups has vastly altered the shapes of the orbitals. For instance, the substitution of a hydrogen for an -OH group has greatly reduced the delocalisation and amount of electron density of the methyl groups in the HOMO compared to the original [N(CH3)4]+ molecule, as apparent from the reduction in size of the MO of the three methyl groups in [N(CH3)3(CH2)OH)]+. This is due to the strong electronegativity effects of oxygen. With regards to the LUMO orbital, it is observed that there is a lot of delocalisation in the [N(CH3)4]+ molecule. A similar situation is seen in the LUMO of the [N(CH3)3(CH2)OH)]+ molecule but a major difference is that, the -OH group, being an electron donating group, has disrupted the symmetry of the original LUMO of the tetramethyl ammonium molecule by establishing electron density sharing between the oxygen, carbon and nitrogen atoms (which was reflected in the charge distribution analysis).
In terms of the HOMO orbital changes in [N(CH3)3(CH2CN)]+, the diminishing of orbitals of the methyl groups and the central part of the molecule is the most striking factor. This is accompanied by the large orbitals associated with the -CN group. The reason is due to the nitrile being a strong electron withdrawing group and therefore, pulling electron density towards itself from the rest of the molecule. In the LUMO of the molecule, it can be seen again that, the orbitals associated with the methyl groups and the central part of the molecule has reduced in size to account for the larger orbitals of the -CN group.
The energy of the orbitals were also changed by the introduction of functional groups. In particular, due to the -OH group, the HOMO and LUMO of the [N(CH3)3(CH2OH)]+ were both raised to a less negative energy, and thus becoming less stabilised due to increasing antibonding character. Because of the increase in energy of the LUMO, the [N(CH3)3(CH2OH)]+ is becoming a weaker acceptor and thus would probably interact less strongly with an anion and thus have a higher melting and boiling point as ion-pair interaction would be less effective. Nevertheless, the introduction of the -OH group may infer hydrogen bonding.
In the [N(CH3)3(CH2CN)]+ molecule, although the HOMO is higher in energy compared to tetramethyl ammonium cation, its LUMO is actually deeper in energy. The reason for the increase in energy for the HOMO is apparent when the orbitals are examined. It can be observed that there is significant antibonding character between the п bond of the -CN group and the p orbital of the neighbouring -CH2 group, leading to the increase in energy. On the other hand, in the LUMO orbital, the original high antibonding character between the diffused orbital engulfing the main body of the molecule and the surrounding methyls in the tetramethyl ammonium compound have now decreased due to reduced orbital size facilitated by the nitrile group. In addition, there is possible bonding interaction between the п* bond of the nitrile and neighbouring methylene group's p orbital. These factors lead to the drop in energy of the LUMO, thereby possibly making this cation better at accepting electrons from anions compared to the previous two molecules.
| Molecule | HOMO-LUMO Energy Gap (a.u.) |
|---|---|
| [N(CH3)4]+ | 0.44632 |
| [N(CH3)3(CH2OH]+ | 0.36304 |
| [N(CH3)3(CH2CN]+ | 0.31865 |
A reduction in the HOMO-LUMO gap can be seen when additional functional groups are introduced. From frontier orbital theory, HOMO-LUMO gap are an important feature of a compound's chemical impact, in that a smaller gap generally implies lower kinetic and thermodynamic stability, as it is not as energetically demanding to add electrons as a high HOMO-LUMO gap. As such, the addition of functional groups would facilitate a better candidate for an ionic liquid, as they would be more likely to form ion pairs.
Conclusion
Three different onoium ions, namely [N(CH3]4]+, [P(CH3]4]+ and [S(CH3]3]+ were examined using computational techniques. Thereafter, additional functional groups, such as an -OH group and a -CN group, were introduced in order to study the effects they may have on the molecule, such as changing the shape and energies of the orbitals. Overall, this investigation has highlighted the potential in which computational studies have in researching potentially novel species of beneficial ionic liquids.
Supplementary Section
It was stated in the optimisation of [N(CH3)3(CH2OH)]+ that manual alterations to the molecule (change of the dihedral angle between the nitrogen and the hydrogen of the hydroxy group, looking down the C-O bond) was required in order to properly optimise the molecule. The data for the failed optimisation as proven by frequency analysis, as well as the reason for the alternation to the molecule that eventually gave the genuine optimisation, are listed below.
Failed Optimisation of [N(CH3)3(CH2OH)]+
The optimisation and frequency analysis was originally performed using the same method (Basis set: 6-31G(d,p), Method: B3LYP), but without distortion of the molecule. Results are listed below.
Optimisation Item Table
Item Value Threshold Converged?
Maximum Force 0.000002 0.000015 YES
RMS Force 0.000000 0.000010 YES
Maximum Displacement 0.000013 0.000060 YES
RMS Displacement 0.000004 0.000040 YES
Predicted change in Energy=-5.682158D-11
Optimization completed.
-- Stationary point found.
It can be seen that a minimum in the potential energy surface has been found. However, this was later prove to be not the correct minimum required but rather, the maximum, i.e. the transition state, by the frequency analysis data:
Frequency Analysis Low Frequencies Table
Low frequencies --- -118.5112 -8.7807 -6.6494 -5.2644 -0.0008 -0.0005 Low frequencies --- 0.0003 129.9524 216.8720 ****** 1 imaginary frequencies (negative Signs) ******
Clearly, the optimisation was not right as the low frequency is massively outside the satisfactory boundaries and also, there is an imaginary frequency present.
Resolving the Problem
3(CH2OH)_animation.gif)
After investigating the imaginary frequency by studying its actual vibration patterns. It can be seen that the problem arises from the hydroxyl group's hydrogen unable to move to a particular side and hence remain in at a maximum as a transition state. Therefore, by changing the dihedral angle between the nitrogen and the hydrogen of the hydroxy group, looking down the C-O bond, one can 'tip' the hydrogen to go one way and hence fall to a minimum in the potential energy surface and thereby the optimisation process finishes at an actual minimum. The alternation was able to resolve the problem and the result in given in the frequency analysis section for [N(CH3)3(CH2OH)]+
References
- ↑ BASF to present BASIL™ ionic liquid process at technology transfer forum, May 10th 2004
- ↑ E. A. Trush, O. V. Shishkin, V. A. Trush, I. S. Konovalovab and T. Y. Sliva, Acta Crystallographica, 2012, E68, o273 DOI:10.1107/S1600536811055024
- ↑ V. S. Mastryukov, J. Struct. Chem., 1976, 17, 1, 69-73 DOI:10.1007/BF00748396
- ↑ L.O.Brockway and H. 0.Jenkins, J. Am. Soc., 1936, 58, 2036-2044 DOI:10.1021/ja01301a063