I began with a 1,5 hexadiene molecule, which was used to determine the low energy minima and transition structure of its corresponding potential energy surface. The reaction is a [3,3] sigmatropic shift but the exact mechanism of the rearrangement has remained largely unclear. It either traverses a stepwise or concerted mechanism but what is clear is that is can go via a boat or chair transition structure. An analysis is therefore done on the transition structures of the Cope rearrangement and to compare the relative energies of the boat and chair transition structures. From the figures below it is shown that the boat transition structures have a slightly higher energy than the corresponding chair transition structures.
Optimizing Products and Reactants
Newman Projection of 1,5 Hexadiene - Figure 1.1
Antiperiplanar
Gauche
Level of Theory: HF/3-21G
I began with isolating the 'antiperiplanar' and 'gauche' structures, shown in Figure 1.1. They were then optimized and the minimum energy found. Optimization involves find the lowest energy position of a nuclei for a given electronic configuration. Due to the Born-Oppenheimer approximation we assume that the electronic configuration will adjust instantaneously with the movement of the nuclei.
Antiperiplanar Conformations Table 1.1
Conformation
Summary Table
Molecular Structure
Point Group
Anti1
C2
Anti2
Ci
Anti3
C2h
Anti4
C1
Gauche Conformations Table 1.2
Conformation
Summary Table
Molecular Structure
Point Group
Gauche1
C2
Gauche3
C2
Gauche4
C1
N.B. Numbering refers to structures outlined in Appendix of Experimental Script.
Based on the results that have been gathered from optimisations of various conformations of 1,5 hexadiene, it is possible to predict the lowest energy conformation. Since antiperiplanar conformers are on average comparable in energy to the gauche conformations, it is difficult to predict without running optimizations, whether one conformer is exclusively lower in energy than the other. From tables 1.1 and 1.2 it is evident that the global minimum is the 'Gauche3' structure with an energy of -231.6927 Hartrees. This can be rationalized by considering orbital overlaps in this gauche form. There is a favorable overlap between π orbital and the vinyl protons[1]. Anti 3,4 and Gauche 1, the CC-eclipsed forms have higher relative energies than CH-eclipsed forms (Anti 1,2 and Gauche 2,3,4 of which Gauche 4 is the highest energy conformer due to the relatively close approach of the 2 hydrogens (2.5Å[2])
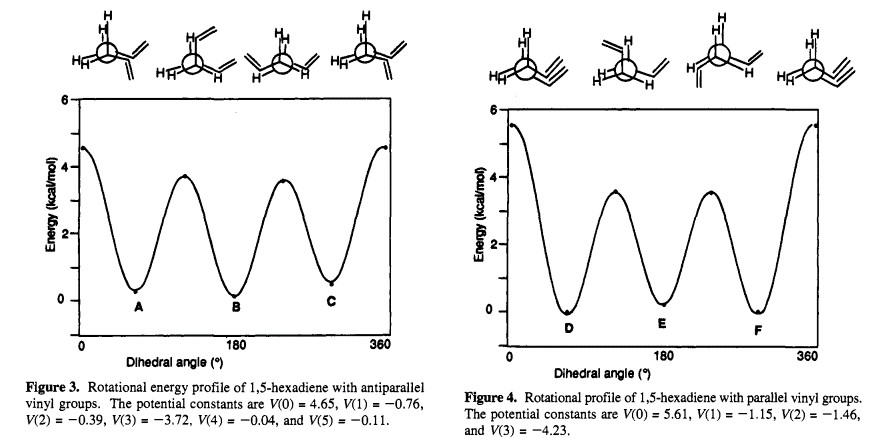
Energy Profile of Parallel and Anti-parallel Conformers of 1,5-hexadiene[3]
An additional consideration is the relative alignment of the vinyl group. However, for the purpose of this investigation, the vinyl groups are aligned parallel.
This shows that there is not a significant difference between having the vinyl group parallel or anti-parallel to each other. Therefore, the relative calculated energies of the cope rearrangement of the hexadiene in one of the two conformers will not be markedly different. Nevertheless, it is still worth noting that these two different conformations exist.
Level of Theory: B3LYP/6-31G
The 'anti2' structure was then reoptimized at a higher level of theory to calculate a more accurate value for its energy. The basis set selected determines the overall accuracy of the optimization. It can be observed from the findings below that the B3LYP/6-31G(d) method gives a slightly lower value for the conformer's energy (-234.611 Hartrees (B3LYP/6-31G(d) vs. -231.693 Hartrees (HF/3-21G)).
Results from Optimizing using B3LYP/6-31G(d) - Figure 1.2
Summary (N.B. Energy is lower than when calculated at HF/3-21G Level of Theory
Molecular Structure
Upon comparing the energies found when using the HF/3-21G level of theory and the B3LYP/6-31G(d) level of theory it is evident that the latter yields a lower energy value. The overall geometry however, appears very similar to each other.
The frequency function in gaussian calculates the second derivatives of energy which allows a stationary point to be characterized. If the output gives a negative eigenvalue or imaginary vibrational frequency this implies that there is a negative curvature at this point, thus corresponding to an energy maximum (Transition structure). Conversely, if the output is real and positive then the geometry of the structure is converging or has converged to an energy minimum. The frequency function of gaussian was then employed to confirm that the optimized structure that was found is indeed a minimum as the optimization may not have found the global minimum but instead the local minimum. There is also a chance we have found a 'saddle point' in which case we have calculated a transition structure rather than a minimum. At a minimum point, we can traverse the potential energy surface in any direction and the energy will always rise, however, for a transition structure, movement in one direction will lead to a reduction in energy which corresponds to the reaction path. Therefore, the frequency calculation must be done in order to identify which stationary point was found.
Upon running the first frequency calculation at the same level of theory (B3LYP/6-31G), the IR vibrations were found and none were imaginary, implying that there is no negative force constant. No imaginary frequencies correlates to the fact that this optimized structure is not a transition structure and is in fact a minimum on the potential energy surface.
Frequency Calculation Findings (After B3LYP/6-31G(d) Optimization) - Figure 1.3
IR Frequencies
Molecular Structure
Output from Gaussian - Figure 1.4
Sum of electronic and zero-point Energies= -234.469204
Sum of electronic and thermal Energies= -234.461857
Sum of electronic and thermal Enthalpies= -234.460913
Sum of electronic and thermal Free Energies= -234.500777
The thermochemistry values from gaussian were compared to the values published[4] with good agreement. The sum of the electronic and zero-point energies correspond to the potential energy at 0K (E=Eelec+ZPE), the sum of electronic and thermal energies at 298.15K and 1 atm pressure (includes translational, rotational and vibrational modes at 298.15K E = E+Evib+Erot+Etrans, the sum of electronic and thermal enthalpies accounts for an additional correction for RT (H=E+RT) and the sum of electronic and thermal free energies accounts for the entropic contribution to free energy.
These values were then recalculated for the same molecule but at 0K using the advanced keywords "Temperature = 0.01(Kelvin)" (as Gaussian does not accept Temperature = 0K). The energies can be predicted even before the calculation is run, and this is because when carrying out an electronic calculation your energy does not depend on temperature. The zero point energy also similarly does not depend on temperature because it is the energy of the electron in the first vibrational state. Both enthalpy and free energy will however, depend on temperature. Therefore, you can see that there is no change in the sum of electronic and zero point energies since by definition the zero point energy is the lowest energy that the conformer can possess (at 0K). Therefore, all the energies converge to the zero point energy value [5].
Output from Gaussian - Figure 1.5
Sum of electronic and zero-point Energies= -234.469204
Sum of electronic and thermal Energies= -234.469203
Sum of electronic and thermal Enthalpies= -234.469203
Sum of electronic and thermal Free Energies= -234.469203
Optimizing Chair and Boat Transition Structures
There has been much debate surrounding which transition structure is reached when 1,5-hexadiene undergoes the cope rearrangement. In this section, the transition structure was optimized and the reaction path obtained to then allow the calculation of the activation energies for the rearrangement via both the chair and boat transition structures.
The transition structure was then "guessed" so that Gaussian can optimize the transition structure. Without the guess, it would be difficult to locate the exact reaction coordinate. To predict the transition structure however is difficult ab initio and therefore an alternative way is to freeze the reaction coordinate using Opt = ModRedundant and minimizing the rest of the molecule before continuing with the transition structure optimization.
Method to Optimize the Chair Transition Structure
Method 1) Obtain 'guess' structure and Optimize at level of theory = HF/3-21G to TS(Berny). The allyl fragment was first optimized and two allyl fragments were placed together to form a guess transition structure.
Figure 1.6
Optimized Allyl
Chair Transition Structure Guess
This will allow the force constant to be computed. When the frequency calculation was run the IR frequencies could be found and only one imaginary was found at -818cm-1. The normal mode corresponding to this imaginary frequency indicates that there is a change in geometry when reactants are transformed into products. From the diagram below it implies that the cope rearrangement is concerted because the bond that is made is formed at the same time the other bond is broken (synchronous process).
Frequency Calculation Findings (After HF/3-21G Frequency Calculation) - Figure 1.7
IR Frequencies
Molecular Structure
Animation of Cope Rearrangement - Figure 1.8
Method 2) Frozen Coordinates
The redundant coordinate editor was used which allows you to create and edit redundant coordinates for use with Gaussian optimization of potential energy scans. The transition structure obtained using this method is similar to the transition structure found by using Method 1).
Frozen Coordinate Method to find Transition Structure - Figure 1.9
Molecular Structure
The bond breaking and bond making distances are fixed to 2.2Å and the bonds are reoptimized again but instead of the "freeze coordinate" function we now use the "derivative function". Since we are not optimizing the transition state a force constant does not need to be calculated. This will allow the bond forming and bond breaking distances to be optimised.
Transition Structure when bond breaking and bond making distances are optimised - Figure 1.10
The bond forming and breaking distances were checked: Bond forming length and bond breaking length are both 2.02Å. It reflects a similar transition structure to the one obtained in method 1.
Method to Optimize the Boat Transition Structure
Method 3) QST2 Method
The boat structure was first optimized using this method. This allows you to specify your reactant and product structures and gaussian will then calculate a transition structure.
Reactants and Products used for QST2 Method using original anti2 conformation - Figure 1.11
In this conformation, gaussian is unable to find the boat transition structure as the conformation of the reactants and products is not conducive to the boat transition structure. Therefore, it fails.
When the chk file was opened the structure shown is similar to the chair transition structure but the two allyl fragments are a bit further apart. It is evident that the QST2 function cannot locate the boat transition structure from these reactants and products.
Transition structure found after running QST2 Method on original anti2 conformation - Figure 1.12
Upon modification of the reactant and product structures to look more like the boat transition structure, the QST2 calculation is run again. Only one imaginary frequency (-840cm-1) is found indicating that a transition structure has been found. An animation is included for illustrative purposes.
New conformations of Reactants and Products for use in QST2 Calculation - Figure 1.13
Animation of Boat Transition Structure (output from QST2) - Figure 1.14
IRC Calculation
Once both the boat and chair transition structures have been found an IRC calculation can be done to compute the reaction path that is traversed during the rearrangement. This is done by calculating the steepest slope between the transition structure to the local minimum on the potential energy surface. The first 50 points were done but this is not sufficient as gaussian indicated:
Output from Gaussian for IRC Calculation (50 steps - Chair Transition Structure) - Figure 1.15
Maximum number of steps reached.
Calculation of FORWARD path complete.
Reaction path calculation complete.
A number of options were available. Firstly, the last point on the IRC was optimized to a minimum however, when the infrared vibrations were checked the minimum was still not reached (not all vibrational frequencies were real and positive), therefore another method was trialled. The minimum may not have been located as the last point on the IRC was still quite far away from the local minimum in which case gaussian may locate an erroneous minimum instead. Therefore, the number of points was increased to 100 points and this led to the calculation of the local minimum.
Output from Gaussian for IRC (100 Steps-Chair Transition Structure) - Figure 1.16
PES minimum detected on this side of the pathway.
Magnitude of the gradient = 0.0001435
Calculation of FORWARD path complete.
Reaction path calculation complete.
In order to determine the activation energies the chair and boat transition structures were both optimised at a higher level of theory (B3LYP/6-31G(d)). A frequency calculation was additionally done in order to obtain the zero-point energies and the activation energy determined.
Output from Gaussian for Chair Transition Structure (B3LYP/6-31G(d)) - Figure 1.19
Item Value Threshold Converged?
Maximum Force 0.000032 0.000450 YES
RMS Force 0.000011 0.000300 YES
Maximum Displacement 0.001375 0.001800 YES
RMS Displacement 0.000672 0.001200 YES
Predicted change in Energy=-3.840311D-08
Optimization completed.
-- Stationary point found.
Sum of electronic and zero-point Energies= -234.414882
Sum of electronic and thermal Energies= -234.408952
Sum of electronic and thermal Enthalpies= -234.408008
Sum of electronic and thermal Free Energies= -234.443125
Output from Gaussian for Boat Transition Structure (B3LYP/6-31G(d)) - Figure 1.20
Item Value Threshold Converged?
Maximum Force 0.000087 0.000450 YES
RMS Force 0.000026 0.000300 YES
Maximum Displacement 0.001096 0.001800 YES
RMS Displacement 0.000436 0.001200 YES
Predicted change in Energy=-1.820299D-07
Optimization completed.
-- Stationary point found.
Sum of electronic and zero-point Energies= -234.398516
Sum of electronic and thermal Energies= -234.392590
Sum of electronic and thermal Enthalpies= -234.391646
Sum of electronic and thermal Free Energies= -234.427342
Output from Gaussian for Reactant(1,5 Hexadiene) (B3LYP/6-31G(d)) - Figure 1.21
Sum of electronic and zero-point Energies= -234.469204
Sum of electronic and thermal Energies= -234.461857
Sum of electronic and thermal Enthalpies= -234.460913
Sum of electronic and thermal Free Energies= -234.500777
Calculations for the Activation energies of the reactions via both Chair and Boat Transition Structures (all at 0K) Level of Theory: B3LYP/6-31G - Figure 1.22
-
Chair Transition Structure
Boat Transition Structure
Reactant Energy (Hartree)
-234.469
-234.469
Transition Structure Energy (Hartree)
-234.415
-234.499
Calculated Activation Energy (kcal/mol)
34.09
44.36
When the same values were taken but at a lower level of theory (i.e. HF/3-21G) there is hardly any change seen in the geometry of the transition state but there is a large difference in the energies obtained. Therefore, it reaffirms that it is initially beneficial to compute transition states at a lower level of theory first to ensure you have the correct geometry and then to reoptimize it again to increase the accuracy of the resulting energy values.
Calculations for the Activation energies of the reactions via both Chair and Boat Transition Structures (all at 0K) Level of Theory: HF/3-21G - Figure 1.23
-
Chair Transition Structure
Boat Transition Structure
Reactant Energy (Hartree)
-231.039
-231.039
Transition Structure Energy (Hartree)
-231.463
-231.451
Calculated Activation Energy (kcal/mol)
266.272
257.424
The calculated values for the activation energies coincide well with the experimental values at 0K and values published by Morokuma et al[6].
Diels Alder Cycloaddition - Butadiene + Ethylene
The Diels Alder Cycloaddition is part of a group of reactions called pericyclic reactions. In a Diels Alder reaction between butadiene and ethylene, the π orbitals of the (usually electron rich) diene overlap with the π orbitals of the (usually electron poor) dienophile to form a new σ bond. The reactions can be categorized into "forbidden" or "allowed" based on the number of п electrons moving in the pericyclic step. The orbital discussion of this reaction begins with the HOMO-LUMO of the diene and dienophile which must come together to produce 4 new bonding and anti-bonding orbitals. In order for the reaction to be classified as "allowed", the HOMO-LUMO of the diene and dienophile must interact on the other hand if its classified as "forbidden" then the HOMO-LUMO are not able to sufficiently overlap because the symmetry of the two are too dissimilar.
The mechanism has been debated over experimentally as there are 3 possibilities - i) a synchronous concerted reaction where the two bonds are formed simultaneously which forms a cyclic aromatic transition state; ii) a two step asynchronous concerted reaction where the two bonds are formed one after the other with two distinct changes in bonding; iii) two step process with with "two kinetically distinct" steps going via a diradical intermediate[7]. When computationally processed, it is commonly agreed that the reaction goes via a synchronous concerted step[8]. The transition state of this Diels Alder reaction involves the diene and dienophile positioned parallel to each other with a perpendicular plane of symmetry running through the middle. Woodward and Hoffman pioneered the selection rules and correlation diagrams to help better understand the mechanism of the reaction. Below are the correlation diagrams for the cycloaddition of 2 ethylenes to form a 4 membered ring and the Diels Alder reaction between ethylene and butadiene. It is evident in the figure below that there must a conservation of orbital symmetry between allowed HOMO-LUMO interactions.
Correlation Diagrams for [4+2] Diels Alder Cycloaddition and [2+2] Cycloaddition - Figure 2.1
The Frontier Molecular Orbital theory requires that the electrons in the HOMO of one the reacting species to 'flow' into the LUMO of the other reacting species. It is sometimes difficult to select which is the donator and which is the acceptor. However, it is usually found that the species with the most п electrons will have a higher energy HOMO. If you compare the molecular orbital diagrams of the two reactants it is illustrated that the HOMO of butadiene is indeed higher than the corresponding HOMO of the ethylene molecule. The red arrows show the interactions that take place during the Diels Alder Reaction. (Shown below)
Correlation Diagram for Diels Alder Cycloaddition between Butadiene and Ethylene - Figure 2.2
In the Diels Alder of butadiene and ethylene the electrons in the HOMO of the butadiene will flow into the ethylene's LUMO. The interaction of the two orbitals will need to interfere constructively so as to lead to the formation of two new sigma bonds. This condition is a part of the Woodward Hoffman rules [9]. This Diels Alder is labelled a [4+2] Concerted Thermal Suprafacial Reaction, as a fragment with 4 electrons (butadiene) is interacting with a fragment with 2 electrons (ethylene) in a suprafacial reaction where both σ bonds are formed on the same face of each of the reactants. Since both HOMO-LUMO pairs conserve orbital symmetry it is thermally allowed according to the Woodward and Hoffmann Rules. Certain orbital arrangements in the transition structure are energetically favorable (symmetry allowed) and some transition structures are energetically unfavorable (symmetry forbidden). Both of the interactions appear to be isoenergetic (shown by red arrows) which means both interactions are equally likely. In this case, we will focus on the HOMO of butadiene interacting with the LUMO of ethylene which will yield a resulting adduct that is antisymmetric with respect to the plane of symmetry.
The HOMO and LUMO orbitals of butadiene were found and are shown below (Figure 2.3).
Molecular Orbitals of Butadiene and Ethylene - Figure 2.3
Molecular Orbitals of Butadiene
HOMO (a) - 1 node
LUMO (s)- 2 nodes
Molecular Orbitals of Ethylene
HOMO (s)
LUMO (a)
The transition structure was 'guessed' once again which is purposely drawn in an envelope type structure maximizing overlaps between the ethylene π orbitals and the π orbitals of butadiene. The transition structure was then optimized using frozen coordinate method to yield the transition structure below.
Level of theory: HF/3-21G
Transition Structure of Diels Alder reaction between Butadiene and Ethylene - Figure 2.4
This can be confirmed to be the transition structure of the Diels Alder reaction as the frequency calculation yields a single imaginary vibrational frequency at -818cm-1. The lowest positive frequency reported is 166cm-1 which is very far from the imaginary vibrational frequency. This means that there is a very definite transition structure at -818cm-1 whereas at 166cm-1 the curvature is not so steep so this point cannot necessarily be classified as a minimum stationary point.
IR Vibrations of Diels Alder Transition Structure between Butadiene and Ethylene - Figure 2.5
IR Vibrations
Molecular Structure
X250px
The molecular orbitals of this transition structure were then found.They can also be classified as either symmetric or antisymmetric with respect to a plane of symmetry.
The modeling of the molecular orbitals has confirmed the theory that the electrons from the HOMO of butadiene (a) will go into the LUMO (a) of ethylene. This reaction is allowed due to Woodward and Hoffmann rules which state that there must a conservation of orbital symmetry[10] which allows for a constructive overlap of the two interacting orbitals.
The HOMO and LUMO orbitals of the transition structure were found.
The Transition Structure link can be clicked to yield a jmol file which can be used to analyze the bond lengths of the newly formed σ C-C bonds. Right click on the screen and click measurements then distances and click the two atoms in which the new C-C bond forms between the distance will appear. They are 0.221nm.
Labelled Bonds in Transition Structure - Figure 2.7
Below is an animation of the vibration that occurs at the Transition state. From the computational analysis done the formation of the two bonds appears to be synchronous. However, there has been much debate about the exact mechanism of the diels alder reaction. Therefore, it cannot be definitively concluded that the mechanism is indeed synchronous. Woodward and Katz[11] postulated that the cope rearrangement was of great relevance to the mechanism of the diels alder as both reactions have similar transition states. In their journal article they mentioned that they found that one bond formed first before the other began to barely form - alluding to an asynchronous mechanism. Yet, on the other hand Dewar[12] reported the presence of a "symmetric cyclic transition state". He additionally completed AM1 computational calculations which revealed that the transition state is indeed synchronous for the butadiene-ethylene diels alder reaction. This finding was further confirmed by additional studies [13][14]
Animation of Diels Alder Reactions between Butadiene and Ethylene - Figure 2.8
Molecular Orbitals of Product - Figure 2.9
HOMO (a)
LUMO (s)
The typical sp3 C-C bond length is 1.54Å and for an sp2 C the C-C bond length is 1.46Å [15].
The bond (2.21Å) that is about to form is shorter than two Van der Waals radii of Carbon which indicates that there is an interaction between the orbitals and therefore the electronic wavefunctions of the reacting species. However, the distance between the two reacting carbons is longer than both a typical sp3 C-C bond length and an sp2 C-C bond length confirming that a bond has not yet formed in the transition structure.
In this reaction we have a very electron poor dienophile (Maleic Anhydride) and an electron rich diene (Cyclo-1,5-hexadiene) which leads to a [4+2] Diels Alder cycloaddition. The reaction can go via 2 transition structures of differing energies. The endo is usually lower in energy than the exo form due to the absence of steric clashes between substituents on the two reacting species.
Molecular Orbitals of Cyclo-1,5-hexadiene and Maleic Anhydride - Figure 3.1
Molecular Orbitals of Cyclo-1,5-hexadiene
HOMO (a) - 1 node
LUMO (s)- 2 nodes
Molecular Orbitals of Maleic Anhydride
HOMO (a)
LUMO (a)
The two interacting species for the purposes of this discussion is the HOMO of the 1,5-cyclohexadiene and the LUMO of the Maleic Anhydride as this has the lowest activation energy and allows for in-phase overlaps between 1,5-cyclohexadiene and Maleic Anhydride. The MOs are shown in figure 3.1 so that visualizing the interaction between these two orbitals is made easier. Since the Maleic Anhydride has electron withdrawing groups on either side of the alkene it is far more electron deficient that an regular ethylene molecule. This will act to make the Maleic Anhydride a better acceptor of electrons from the 1,5-cycloehexadiene. A diagram of the molecular orbital interactions is shown below.
Schematic Diagram of Endo and Exo Conformations - Figure 3.2
Exo and Endo Transition Structures - Figure 3.3
Exo Transition Structure
Endo Transition Structure
Energies of Endo and Exo Transition Structure (0K) (HF/3-21G) - Figure 3.4
Endo
Exo
-605.414905 Hartree
-605.518387 Hartree
Endo TS IR Vibrations
Exo TS IR Vibrations
The IR vibrational frequencies found for both the endo and exo transition structures confirm that they are indeed transition structures (single imaginary vibrational frequency). However, by doing ab initio calculations at the HF/3-21G level, the energies found did not match the theory, so the transition structures were reoptimized again at a higher level of theory (B3LYP/6-31G(d)) and the energies obtained are displayed below. Again it is clear that although the geometries of the transition structures obtained at the HF/3-21G level of theory and B3LYP/6-31G(d) level of theory the energy are dramatically different which reaffirms that it is useful to use a lower level of theory to do an initial optimization and a higher level of theory to improve it.
Energies of Endo and Exo Transition Structure (0K) (B3LYP/6-31G(d)) - Figure 3.5
Endo
Exo
-612.502141 Hartree
-612.498013 Hartree
Endo TS IR Vibrations
Exo TS IR Vibrations
Again, it is confirmed that these are in fact transition structures due to their single imaginary vibrational frequencies. It is evident that the endo transition structure is of a lower energy than the exo transition structure which is expected as the endo product is usually preferred. This can be explained by the fact that there is more strain due to sterics in the exo transition structure making this reaction pathway slower due to a higher activation energy. The exo form is more strained because there is a greater steric clash between the hydrogens from the cyclohexadiene and the oxygens on the maleic anhydride. The preference for the endo conformer is explained further in Molecular Orbital Discussions.
Animation of Endo and Exo Transition Structures - Figure 3.6
Endo
Exo
Molecular Orbital Discussions
Molecular Orbitals of Exo and Endo Transition Structures - Figure 3.7
HOMO
Endo
Exo
LUMO
Endo
Exo
From the molecular orbital modeling above, it is shown that the HOMO of the Endo transition structure has many nodes. When examining the nodal properties of the HOMO between the -(C=O)-O-(C=O)- fragment it is evident that there is some constructive overlap in the endo form which is less evident in the exo form. It can also be noted that in the HOMO of the exo conformer there are large molecular orbitals which do not overlap constructively with the MOs nearby and hence there are more clear-cut nodes than in the endo conformer. The extra overlap can be attributed to the secondary orbital overlap that explains the stereospecificity for the endo conformer over the exo conformer.
Secondary Orbital Overlap
The in-phase requirement for the reaction to be thermally allowed is considered a first order orbital overlap and this determines the stereospecificity of the reaction. Another interaction that needs to be considered especially in the discussion of exo- and endo- transition structures is the secondary orbital overlap[17] which determines the regio- and stereoselectivity of the reaction. Experimentally it is observed that there is an overwhelming 99:1 ratio for the endo product even though the energy difference between the two is only 3 kcal/mol which agrees well with the energy difference found at the B3LYP/6-31G(d) level: 2.590 kcal/mol. There is an overall preference for the endo product due to the secondary orbital overlaps that are present in the endo form but not in the exo form[18]. This is exemplified in Figure 3.2 where you can see the relevant overlaps in the exo and endo conformations.
An IRC calculation was attempted to see the steps between the Transition Structure and the product however, the system was too large for gaussian to handle and the calculation took too long to compute.
Further Discussions
These computational calculations do not necessarily reflect experimental findings as they tend to invoke approximations so that a wide range of reactions can be modeled. The first of these approximations is the Born-Oppenheimer approximation which fixes the positions of the nuclei to only allow for the electron distribution to be investigated. This simplifies the Schrodinger equation greatly so that it is not necessary to treat each atom quantum mechanically. We have to simplify the system at hand by ignoring individual interactions between nuclei and electrons and instead average the electron-electron interactions and electron-nucleus interactions. We cannot completely ignore the interactions as this would lead to an erroneous calculated energy. Since electrons are much smaller than the nuclei, we can assume that the electron distribution will change instantaneously when the nuclei move. Another limitation of computational methods of analyzing transition structures is the lack of steric consideration which may be a dominant factor in deciding which orientation the two reactants react in. As the user has to align the transition structure manually, it is imperative that there is some idea of the conformation of the transition structure. However, if there is a reaction where the transition structure is difficult to observe experimentally, then computing it with gaussian would be difficult. Nevertheless, using the QST2 function could work but yet again, the reactants and products must be oriented to appear similar to the transition structure and without a rough idea of what the transition structure looks like this could prove difficult as well. However, the benefit of doing these calculations computationally is that we are able to locate and calculate the energies of transition structures as this is difficult to do experimentally (for reactions such as the Diels-alder where the transition structure is well known).
References
↑ B. W. Gung, Z. Zhaohai, and R. A. Fouch, J. Am. Chem. Soc., 1995, 1783–1788.
↑ B. W. Gung, Z. Zhaohai, and R. A. Fouch, J. Am. Chem. Soc., 1995, 1783–1788.
↑ B. W. Gung, Z. Zhaohai, and R. A. Fouch, J. Am. Chem. Soc., 1995, 1783–1788.
↑K. Morokuma, W. T. Borden, and D. A. Hrovat, J. Am. Chem, 1988, 4474–4475
↑F. Bernardi, A. Bottoni, M. J. Field, M. F. Guest, I. H. Hillier, M. A. Robb, and A. Venturini, Journal of the American Chemical Society, 1988, 110, 3050–3055 DOI:10.1021/ja00218a009
↑M. Torrent, M. Duran, and M. Sola, Scientia Gerundensis, 1996, 123–131
↑ R. Hoffmann and R. B. Woodward, Journal of the American Chemical Society, 1965, 87, 2046–2048. DOI:10.1021/ja01087a034
↑ R. Hoffmann and R. B. Woodward, Journal of the American Chemical Society, 1965, 87, 2046–2048. DOI:10.1021/ja01087a034












































































{kind=link}