This computational exercise investigates the time evolution of a Lennard-Jones fluid in different ensembles: the canonical ensemble [N,V,T] and the Gibbs ensemble [N,P,T] in the later sections. The need for ensembles arises due to the difficulties of determining a vast number of particles' thermodynamic properties. And in order to justify the use of ensemble averages, the sample size needs to be sufficiently large as described by the Ergodic hypothesis, which states that as time tends to infinity (or that all possible microstates are being simulated), the average of such system can be treated as the time average.[1]
Figure 1: Ergodicity
The fundamental basis for Molecular Dynamics Simulations lies in capturing the time evolution of a series of interacting particles. Running the MD simulations is a solution to the challenge to solving a 'many bodies' problem, by using numerical integration and discretization, which is the transfer of continuous functions into solvable, discrete components. To perform the molecular dynamics simulation, several factors need to be specified first in the input scripts.
Mass and atom types
The potential function to describe atoms' interactions.
Initial velocities and positions
Time step at which the thermodynamic and physical conditions will be updated.
The ensemble ( a collection of independent systems that abide by identical boundary conditions and properties. In this case, the ensemble is NVE, fixing the total number of particles, the volume and the total energy of the system).
An algorithm is required to propagate the positions and velocities in the time interval using numerical integrators, which differ in the number of higher order terms included as well as whether they include previous time step in the variable time step schemes. It is also noteworthy to point out that the time step used must correlate with the fastest motions in the systems to capture the changes of the system and ensure that the integration is stable. The physical quantities resulted from each system in the ensemble are then averaged to give the overall physical property of the system. The quality of results depends on the number of systems being sampled, as Ergodic hypothesis applies (the average of an infinitely large system's physical properties can be treated as the time average of the system). In this experiment, the Velocity Verlet Algorithm was used to model the behavior of the classical harmonic oscillator. The initial conditions are specified, then after the time step is implemented, the positions and the half-step velocities are updated and that the forces acting on the atoms are calculated.And finally, during each time step, the position and velocity are integrated to form a trajectory. Finally, specific microscopic properties such as the radial distribution function and the velocity autocorrelation functions will be derived from every phase of the Lennard-Jones fluid, providing a link between the microscopic-nature of the simulations with macroscopic thermodynamic properties.
In summary, after choosing the appropriate initial parameters and creating the simulation box in form of an ordered lattice, the simulation will proceed to collapse and melt the lattice to produce the necessary fluid state from which thermodynamics properties can be derived; the accuracy of which will be determined predominantly by the sample size and time step, as the following sections will examine in greater detail.
Modelling the behavior of the classical harmonic oscillator
As an approximation to simulating N-bodies of atoms in the systems, the atoms are treated as small, hard spheres obeying Newtonian laws in their trajectories, velocities and positions. This is incorporated into the Velocity-Verlet algorithm, which uses numerical integration to find the updated positions and velocities at some time, dt, later. Given the possible non-uniformity of position and velocity curves, the integration procedures must include discretization to calculate smaller areas under the curve and then summing these areas. As a consequence, we can identify immediately the sources of errors with the algorithm, that is the size of timestep, dt, or at which the scale at which discretization is being applied, as well as the number of terms allocated to the Taylor's expansion, shown below with the error term:
Figure 2: Taylor Expansion used in the Verlet algorithms
This is termed as the round-off error and as such, any Verlet simulations must ensure that the final term be relatively negligible in comparison to the other terms in the expansion.
The positions of the classical harmonic oscillator, updated positions using the Velocity-Verlet algorithm and the total energies are described below:
HO Analytical Position Function
Velocity Verlet Algorithm's
Position Function
Total Energy
where A is the amplitude, ω is the angular frequency and φ is the phase of the oscillation. The resulting analytical position is sinusoidal with respect to time and is shown to be in close agreement with the positions generated from the Velocity Verlet Algorithm. As shown through the error outputs (defined as the absolute difference between the classical solution and the Velocity Verlet Algorithm outputs), the error displays a series of increasing maxima over time. And finally, the energy outputs are defined by summing the kinetic energy and the potential energy of the physical system at the given time. The kinetic term uses the velocity derived from the Verlet Algorithm and that the potential energy follows a harmonic oscillator where, PE = 0.5kx2.Calculations are done on HO.xls file, where the initial timestep used for all calculations was 0.1.
Analytical Position Output with 0.1 Time Step
Errors (Difference between Velocity Verlet Algorithm and Classical Solution)
with 0.1 Time Step
Total Energy with 0.1 Time Step
Gallery 1: Comparison between the Velocity-Verlet algorithm outputs with the 1D Harmonic Oscillator model.
The Velocity Verlet is a type of numerical integration algorithm, and so there is an inherent truncation error from using too few terms in the Taylor expansion in addition to the integration error from using larger time step instead of incremental time steps. As a result, it is expected that the error increases with time, as each error term accumulates over time due to the propagation of error over all the time steps involved in the simulation (the cumulative errors associated with the previous time-steps are added to the current error associated with the current timestep).
Maxima on the Error Plot
0.00076
0.0020
0.00330
0.00457
0.00591
Corresponding time
2.100
5.000
8.000
11.200
14.250
The fitted plot has near absolute linearity, R2 = 0.9998, suggesting that there is minimal error in the relationship between the plot maxima and time. Errors in the Velocity Verlet algorithm can also be manifested in the energy output given that the total energy should remain constant given the law of energy conservation. It is observed than the time step and errors' magnitude has positive correlation and increasing the time step beyond 0.2 results in the total energy changes more than 1% during the simulation.
Time Step
0.1
0.2
0.3
0.4
0.5
Maxima
0.500
0.500
0.500
0.500
0.500
Minima
0.4987
0.4950
0.4888
0.4800
0.4687
Percentage Change
0.2%
1%
2.25%
4%
6.25%
Choosing smaller time steps, showing that for time step below 0.2, the total energy does not change by more than 1% give the following percentage differences in total energy over time:
Time Step
0.12
0.14
0.16
0.18
0.19
Maxima
0.5000
0.5000
0.5000
0.5000
0.5000
Minima
0.4982
0.4976
0.4968
0.4959
0.4955
Percentage Change
0.36%
0.49%
0.64%
0.81%
0.90%
From the calculations, it is evident that the time step cannot exceed 0.2 to result in less than 1% change in the total energy fluctuations. This is important because minimizing this discrepancy allows the simulation to be more realistic (abiding by energy conservation).
Atomic Forces and the Lennard-Jones Interaction
The single Lennard-Jones interaction (12-6 interaction) and the sum of all pairwise interactions are described by the following equations. The 12-6 potential is characterized by a parabolic function, in which the potential tends to zero as separation tends towards infinity and that the potential tends to infinity at distances smaller than the equilibrium separation.
The separation between the atoms engaging in a single Lennard-Jones interaction is σ.
The force acting on the atom is determined using the derivative of the potential energy, and in this of a zero Lennard-Jones potential, the force is equal to 24ε/σ. To solve for the force at this separation,
Figure 5. Definition of Force as per Newtonian law
Taking the derivative with respect to the Lennard-Jones potential,
Figure 6. Force as described with Lennard-Jones potential.
The equilibrium separation is found by finding the minimum of the Lennard Jones potential curve, when the force is equal to zero. Using the same equation as shown above, the separation is solved:
Figure 8. The equilibrium seperation
The well depth of the parabolic Lennard-Jones potential is the distance from zero potential to the Lennard-Jones interaction at the equilibrium position. Substituting the equilibrium separation into the Lennard Jones potential, the absolute wall depth is ε.
Evaluation of the following integrals, where σ=ε=1 :
The integral evaluated is equal to -0.0252.
The integral evaluated is equal to -0.0082.
The integral evaluated is equal to -0.00329.
Periodic Boundary Conditions
The number of water molecules in 1ml of water under standard conditions can be determined using the density of water at 298 K and 1 atm, which is 0.997 g/ml. Solving for the number of moles of water molecules and then converting the quantity using Avogadro's number (6.022x1023 mol-1), the number of water molecules is 3.345 x 1022. Similarly, the volume that 10000 water molecules occupy under standard conditions are calculated in the identical method, where V = 2.997 x 10-19 ml3.
Periodic boundary conditions are applied to the system of bulk liquid as to ensure an infinitely large system, by partitioning the system into individual unit cells. The boundary conditions ensure that once an object has passed through one face of the simulation unit cell, it should re-enter based on the vector at which it moves along. For a cubic unit cell running along (0,0,0) to (1,1,1) in a single time step and initial atom at position (0.5, 0.5, 0.5), the final position of the atom is determined using the vector sum of the initial position and the vector at which it moves along. This corresponds to (1.2, 1.1, 0.7) and applying the boundary conditions, the atom ends up at (0.2, 0.1, 0.7) of the simulation box.
Reduced Units
Distance,
Temperature,
Potential Well,
Scaling factors are used to ensure that the values being used for the Lennard-Jones interaction are simplified in the input scripts. The distance, r, is being converted by dividing the value with σ or the separation producing zero Lennard-Jones interaction. Energy is divided by ε or the depth of the potential well. And finally, temperature is expressed as the thermal energy divided by ε.
For argon which has σ = 0.34 nm and ε/KB = 120K, the real units of separation is 1.088 nm if the reduced unit is 3.2 nm. The wall depth is 0.997 kJ mol-1 and that the reduced temperature, 1.5, in real units is equal to 180K.
Equilibration
This section of the computational exercise seeks to determine the thermodynamic properties of the system based on the initial velocities and time steps used. For the purpose of this investigation, five text input scripts are created. All of the scripts will simulate the same system but each will have an associated unique time step. The five time steps being investigated are 0.001, 0.0025, 0.0075, 0.01 and 0.015. Simulations are conducted on the Imperial High Performance Computing (HPC) systems and through LAMMPS (Large-scale Atomic/Molecular Massively Parallel) Molecular Dynamics Simulator. The LAMMPS program uses a Verlet list to maintain information of all particles during the molecular dynamics simulations.
Creating the Simulation Box
The requirement for periodic boundary conditions arise from the fact that the number of cells is finite in the simulations, and as a result, the behavior of the particles near the edges need to be accounted for. Periodic boundary conditions allow any particles that travel past the edges to be wrapped back to the cell from the opposite side, thereby effectively eliminating boundaries.
For any simulations to run, the initial conditions of all the atoms in the systems need to be specified. In this case, an aqueous system differs from a crystalline system in that there is no transnational symmetry of particles/atoms. Yet, giving atoms random starting coordinates in simulations would incur problems relating to computational resources. In particular, scenarios where atoms are found to be too close together or overlapping with each other generates an infinitely repulsive Lennard-Jones interaction. By restricting the starting atom coordinates to realistic cases, less resources are required for running the simulations.
Therefore, the atoms will be placed accordingly to the arrangement of lattice points of a cubic lattice. The lattice will then collapse and the atoms will rearrange themselves into a more realistic configuration, assuming that the allocation of time is sufficient. Taking one of the input script, 0.0025.in, the following command line specifies the number density for the type of lattice (sc = simple cubic lattice) is as follows:
lattice sc 0.8
And then, looking at the output file corresponding to 0.0025.in, the following command line specifies the distance between the points in the lattice in three dimensions.
Lattice spacing in x,y,z = 1.07722 1.07722 1.07722
For the above command lines, the total volume is 1.25001 and that given there is only one lattice point per unit volume for a simple cubic lattice, the number density is 0.8. For a face-centered cubic lattice with number density of 1.2, the side length of the cubic unit cell is 1.4938. This is derived from the fact that a fcc unit cell possesses four lattice points per unit volume, solving for 1.2 = 4/l3 gives l = 1.4938.
To create a geometrical box to accommodate the lattice points and therefore the simulation box, the following command lines are used:
region box block 0 10 0 10 0 10
create_box 1 box
create_atoms 1 box
The lines are instructions for LAMMPS to create a cube extending 10 lattice spacings in three dimensions; and within that box, only one type of atom will be present. Given the previous command line specifying a simple cubic lattice with number density of 0.8, the result is 1000 simple cubic unit cells. The create_atoms instructs LAMMPS to populate the 'box' with atoms of type 1. If the earlier command lines had specified a fcc type lattice, and follow the same subsequent command lines, we can expect the output to be a box with 1000 unit cells with 4000 atoms, given that each unit cell in fcc lattice has 4 lattice points. It is also important to note that the initial simulation lattice must be suitable with respect to the number of time steps allocated if any equilibration is desired. This is because by not minimizing the initial energetic state of the system, the system of atoms will use the time-steps trying to reach a new stable configuration and therefore there may not be enough time to equilibrate.
Setting the properties of the atoms
The properties of the atoms are defined on a 'per-type' basis and is done using the following commands,
The 'mass' command line set the mass for all atoms for the specified atom type. '1' refers to the atom type and 1.0 refers to the per-type mass value set forth. Next, the pair_style command describes a pair potential and lj/cut computes the 12/6 Lennard-Jones potential, with the cut-off internuclear distance at 3.0 (in reduced unit). The last command line, pair_coeff describes the pairwise force field coefficient for the pair of atom types; the asterisks refer to all atom pairs present in the lattice, while 1.0 value that follows describes the force field coefficients.
As mentioned previously, the Velocity Verlet algorithm in the Molecular Dynamics simulation is used and therefore the initial positions and initial velocities need to be specified before the simulation can be carried out. The determination of the atoms' initial random velocities is done using the Maxwell-Boltzmann distribution, and therefore the temperature is needed to calculate the distribution.
The following command lines are found in the input files:
### SPECIFY TIMESTEP ###
variable timestep equal 0.001
variable n_steps equal floor(100/${timestep})
variable n_steps equal floor(100/0.001)
timestep ${timestep}
timestep 0.001
### RUN SIMULATION ###
run ${n_steps}
run 100000
By defining a variable and making subsequent command lines dependent on it, the number of manual inputs required to run the simulation can be reduced significantly. As shown above, by defining timestep at the first command line, the command would cascade down the input script without the need for manual inputs every time the time step needs to be called out. From a technical perspective, this also ensures consistency in the input script and minimize errors caused for manual intervention. This differs from the command line below, where:
timestep 0.001
run 100000
In which case, two manual inputs are required (defining the time step and also the number of runs required). As compared to the first set of command lines, by changing the time-step the other input lines will follow automatically.
Visualizing the trajectory
The trajectory files contain a 'dump' command which captures the snapshot of atom positions every N timesteps, and that the file defines a set interval at which the positions of the atoms are recorded in the simulation. VMD, a molecular visualization program, is used to visualize the change in atomic positions over time, The images below illustrate the different visualizations of the 0.015 time-step experiment's output.
Description
VMD Representations
Line Representation
at t = 0.0
VMD Representation
at t = 0.0 for a
simple cubic lattice
Orthographic Display
Illustrating Periodic Boundary Condition at t1
Illustrating Periodic Boundary Condition at t2
As per molecular dynamics simulation, the simulation box is also represented by a periodic boundary condition to satisfy the requirement of having an infinitely large system for ergodic hypothesis to be valid. Running the trajectory visualization, where t2>t1 in time, it can be seen that the atoms trespass the boundary in the y-direction, only to return to the simulation box from the opposite face.
Checking Equilibration
In Molecular Dynamics simulation, the system reaches equilibrium when the averaged thermodynamic quantities become constant and fluctuate about the same average value. Each of the fluctuation can be described using Gaussian distributions. A series of output files will be compared according to the time-steps specified in the earlier input text files to check if equilibrium has been reached.
0.001 Time-Step
0.0025 Time-Step
0.0075 Time-Step
0.01 Time-Step
0.015 Time-Step
Gallery 2: The variation of total energy (in absolute value) versus time for a range of time-step experiments.
0.001 Time-Step
0.0025 Time-Step
0.0075 Time-Step
0.01 Time-Step
0.015 Time-Step
Gallery 3: The variation of temperature (in reduced unit) versus time for a range of time-step experiments.
0.001 Time-Step
0.0025 Time-Step
0.0075 Time-Step
0.01 Time-Step
0.015 Time-Step
Gallery 4: The variation of pressure (in reduced unit) versus time for a range of time-step experiments.
For the 0.001 time-step experiment, equilibrium is reached as shown by the constant Total Energy value after t=0.2. Given that the unit for Total Energy is in reduced unit (shown below), the variation of energy values in the fluctuations is not significantly large. Taking the maximum and minimum values of the last 20 total energy values of the 0.001 Time-Step experiment (-3.183815 and -3.184684), the difference is 1.439 x 10-24 J. Similarly, the temperature and pressure for all time-step experiments stabilize in the course of the stimulation.
Time-Step Experiment
Average Total Energy Value
(Calculated using the last 20 points)
0.001
-3.1842794
0.0025
-3.1837034
0.0075
-3.1823043
0.0100
-3.1815204
0.0150
-3.1474706
Table 1. The averaged total energy values in different time-step experiments
Figure 9. Comparison of the total energy fluctuations over time between time-step experiments.
From the comparison between all the time-step experiments, it is evident that the most accurate results tend to the one produced from the experiment using the lowest time step. In this case, both 0.001 and 0.0025 time-step experiments produce identical total energy values at the end of the simulation. The deviation becomes larger from using experiments of larger time-steps. It is reasonable to conclude, therefore, that the best compromise is to utilize 0.0025 time-steps to produce accurate results yet, having sufficiently large time-steps.
The errors associated with the time-step experiments are the result of truncation, where the additional terms as part of the Taylor's series becomes substantially important to the result when the time-steps are larger. Due to the need to conserve computing resources, truncation and the cutoff internuclear distance for the Lennard-Jones interaction are required, which is justified due to negligible forces between particles that are far apart. As a consequence, the conservation of energy is violated due to the exclusion of long-range order interactions.
Both 0.0075 and 0.01 time-step experiments are more inaccurate compared to simulations with smaller time steps (the simulation fixed the number of terms to be included in the Taylor expansion). Finally, the 0.015 simulation is entirely inaccurate given that there is no conservation of energy, as shown above. This observed behaviour is termed as 'exploding' and result in impactful atomic collisions, as the large time-step would force the particle to overshoot and result in a higher-than-expected total energy.[2] And with this erroneous total energy, the simulation continues and cause the energy to increase indefinitely, as shown.
Running Simulations Under Specific Conditions
For the subsequent simulations, a different ensemble environment will be employed to investigate the behaviors in an [N,P,T] ensemble. The ten input scripts for this exercise are characterized by 5 variable temperature for each of the two variable pressures determined from the minimum and maximum values of the pressure fluctuations in the previous section. In this case, the five temperatures (in reduced units) chosen are 1.5, 2.0, 2.5, 3.0, 3.5 and 4.0. And the two pressures values (in reduced units) are 2 and 3. The values for the pressure are determined using the outputs from the Equilibration section, where it is evident that from all time-step experiments, the pressure fluctuates from 2 to 3. The temperatures are chosen to be above the critical temperature of TC = 1.5, where there is a 2-phase coexistence observed in the system of Lennard-Jones fluid (TC represents the temperature at which the vapour of the Lennard-Jones fluid cannot be liquefied, irrespective of the pressure applied to the system). Finally, all simulations are performed using the 0.0025 time step determined using the equilibration exercise in previous section.
Simulating Thermostat and Barostat Conditions
To ensure that the thermostat condition is constant throughout the simulation, there needs to be constant readjustment of the kinetic energy of the system to ensure that the instantaneous temperature can correlate to the pre-set temperatures in the input files for NpT ensemble simulations. The equipartition theorem states that each degree of freedom in a system contributes 0.5KBT of energy. Similarly, the kinetic energy is the sum of all terms from the ensemble's atoms, and so:
Figure 10. Sum of all atomic kinetic energies
By multiplying the velocity of each individual atom with a constant, the temperature can be changed (as a result of proportionality on either side of the equation), where T is the instantaneous temperature needs to be converted to the specified initial temperature shown on the right:
Figure 11. Kinetic energy with converted temperature
Solving the two equations, give a relationship between the pre-set temperature and the instantaneous temperature. And from this, the constant required to multiply the velocity can be determined:
Figure 12. The derived temperature correction term
To ensure that the pressure is constant, the size of the simulation box has to changed at every time step; this indicates that volume is not kept constant in the ensemble as the temperature fluctuates during each simulation.
The Input Scripts
To ensure that the simulation ensures constant control in temperature and pressure, the following command line is included in the script:
fix npt all npt temp ${T} ${T} ${tdamp} iso ${p} ${p} ${pdamp}
By specifying ${T} twice, it ensures that the desired starting and final temperatures in the simulation are equal (the same applies for ${p} with respect to pressure). The subsequent command lines then measures the thermodynamic properties of the system at the end of the simulation:
### MEASURE SYSTEM STATE ###
thermo_style custom step etotal temp press density
variable dens equal density
variable dens2 equal density*density
variable temp equal temp
variable temp2 equal temp*temp
variable press equal press
variable press2 equal press*press
fix aves all ave/time 100 1000 100000 v_dens v_temp v_press v_dens2 v_temp2 v_press2
run 100000
The first line, thermo_style, allows the outputs of the simulation to determine which thermodynamic properties to display. 'Thermo_style custom step etotal temp press density' will only produce a line with numeric values that are instantaneous and calculated on the exact timestep. The subsequent six command lines then define the variables associated with each thermodynamic property being measured, as well as values that will be used for the calculation of the standard error, which depend on the standard deviation and hence the need for the squares.
The time-averaged quantities are represented by the command line, and they specify the number of time steps that will result in the averaging process for each thermodynamic property.
The general syntax for the fix ave/time command is 'fix ID group-ID ave/time Nevery Nrepeat Nfreq value1 value2 value3 ....keyword args...' 100 is the Nevery value in this command line, and represents the number of time steps elapsed before an input value is used to contribute to the average (in our case, every hundredth value will be used to compute the final average). Nfreq refers to the number of time-steps that must have elapsed for the averaging process to occur. Finally the keywords, v-dens, v_temps, v_press, v_dens, v_temp2 and v_press2, are the variables that will undergo the averaging process. In total, the time used for the simulation is the product between the time-step and Nfreq, which is 100000*0.0025=250 seconds.[3]
Plotting the Equations of State (Comparing Simulation Outputs with Ideal Gas Law)
The outputs for the number density versus temperature for the isobaric-isothermal ensembles are shown below. Converting the Ideal Gas Law to include density results in the modified expression:
Figure 13. Modified Ideal Gas Law
Comparison of Densities Calculated in the [N,P,T] Ensemble at P=2
Comparison of Densities Calculated in the [N,P,T] Ensemble at P=3
From the Molecular Dynamics simulations, it is evident that the errors associated with the densities and temperatures are not large; 0.00433 and 0.0385 respectively for P=2 system, 0.00423 and 0.0386 respectively for P=3 system. In both simulations, the MD resulting averaged densities are consistently lower than the predictions from the ideal gas law at every temperature. Deviations from the ideal gas law can be seen to decrease with increasing temperatures (applicable to both simulations at P=2 and P=3), and this correlates directly to the requirements for ideal gas law to be valid. To analyze why the deviations occur, it is important to first note the assumptions made with ideal gas law:
Collisions between the atoms (which are considered as small and hard spheres) are elastic and therefore frictionless.
Atoms' trajectories and velocities follow Newtonian laws.
The sum of individual volume occupancy of each atom is negligible compared to the total volume of the system.
There is a random distribution of atoms, moving with a distribution of speeds due to the lack of attractive or repulsive forces to direct them in specific directions.
Low densities or high temperatures must be sustained.
From these assumptions, it becomes evident that the ideal gas law does not tailor to systems with repulsive and attractive forces acting on each atom, which happens to be the cornerstone for simulating Lennard-Jones fluid. In the ideal case, the potential energy of any given pair of atoms should not exist, as the volume-assumption asserts that the total volume occupancy of all atoms is negligible compared to the total volume of the system. However, as shown by previous sections, not only do the atoms possess pairwise interactions, these interactions extend to longer range order and hence the repulsive and attractive terms of Lennard Jones potential need to be taken into consideration. The observation that at higher temperatures, these discrepancies decrease, are due to the fact that atoms are imparted with greater kinetic energy, and by law of energy conservation, this suggests that the potential energy term becomes increasingly less dominant (this allows the system to tend to the idealized scenario).
Taking the density values at T=2.00 (past the critical temperature), the discrepancy with ideal gas law at P=2 is 46.8% while the discrepancy at P=3 is 61.5%. This is because at increased pressure (higher density), the short-range order of the Lennard-Jones interaction becomes even more pronounced, leading to greater dominance of potential energy in calculating the total energy. Evidently, P=3 would result in a greater deviation from the idealized case as compared to P=2.
Heat Capacity Calculations
In statistical thermodynamics, the heat capacity describes the fluctuations of internal energy in the canonical ensemble. The equation for heat capacity in the canonical ensemble is described below:
Figure 14. Heat Capacity as a description for variance.
By nature of the equation, the heat capacity is not an ensemble average. The following simulations seek to derive the relationship of the heat capacity with respect to temperature in the N,V,T ensemble. Temperatures being tested are 2.0, 2.2, 2.4, 2.6 and 2.8 with densities equal to 0.2 and 0.8. As previously, the timestep allocated to the simulation is 0.0025 to ensure accuracy and completion. As an example, the input script for ρ = 0.2 and T=2.2 can be found below:
###NEW VARIABLE FOR INITIAL DENSITY###
variable initialD equal 0.2
''<u>>The first step is to define the new variable required for the simulation, which will need to be called out in the subsequent steps. In this case, the initial density is 0.2.</u>''
### DEFINE SIMULATION BOX GEOMETRY ###
lattice sc ${initialD}
region box block 0 15 0 15 0 15
create_box 1 box
create_atoms 1 box
''<u>>As before, the simulation box commands do not change, with the exception of including a density variable.</u>''
### DEFINE PHYSICAL PROPERTIES OF ATOMS ###
mass 1 1.0
pair_style lj/cut/opt 3.0
pair_coeff 1 1 1.0 1.0
neighbor 2.0 bin
### SPECIFY THE REQUIRED THERMODYNAMIC STATE ###
variable T equal 2.2
variable timestep equal 0.0025
### ASSIGN ATOMIC VELOCITIES ###
velocity all create ${T} 12345 dist gaussian rot yes mom yes
### SPECIFY ENSEMBLE ###
timestep ${timestep}
fix nve all nve
''<u>>The first step is to define the new variable required for the simulation, which will need to be called out in the subsequent steps. In this case, the initial density is 0.2.</u>''
### THERMODYNAMIC OUTPUT CONTROL ###
thermo_style custom time etotal temp press
thermo 10
### RECORD TRAJECTORY ###
dump traj all custom 1000 output-1 id x y z
### SPECIFY TIMESTEP ###
### RUN SIMULATION TO MELT CRYSTAL ###
run 100000
unfix nve
reset_timestep 0
''<u>>The nve ensemble is 'unfix' given that the crystal needs to melt to achieve a liquid/gaseous phase. </u>''
### BRING SYSTEM TO REQUIRED STATE ###
variable tdamp equal ${timestep}*100
fix nvt all nvt temp ${T} ${T} ${tdamp}
run 100000
reset_timestep 0
unfix nvt
fix nve all nve
''<u>>NVT ensemble is fixed as this is the ensemble at which the system needs to operate in. Once the system is brought to its required state, the NVT ensemble is unfixed. </u>''
### MEASURE SYSTEM STATE ###
thermo_style custom step etotal temp press density atoms
variable dens equal density
variable dens2 equal density*density
variable temp equal temp
variable temp2 equal temp*temp
variable press equal press
variable press2 equal press*press
variable etotal equal etotal
variable etotal2 equal etotal*etotal
variable atoms equal atoms
fix aves all ave/time 100 1000 100000 v_dens v_temp v_press v_dens2 v_temp2 v_press2 v_etotal v_etotal2
run 100000
''<u>>New variables for E and E^2 needs to be called out, by referencing to the thermodynamic outputs from the simulation. As previously, the averaging process and the number of samples we used for averaging do not change, but E and E^2 need to be referenced here to calculate the variance in subsequent steps. </u>''
variable avedens equal f_aves[1]
variable avetemp equal f_aves[2]
variable avepress equal f_aves[3]
variable aveetotal equal f_aves[7]
variable aveetotal2 equal f_aves[8]
variable errdens equal sqrt(f_aves[4]-f_aves[1]*f_aves[1])
variable errtemp equal sqrt(f_aves[5]-f_aves[2]*f_aves[2])
variable errpress equal sqrt(f_aves[6]-f_aves[3]*f_aves[3])
###MEASURE HEAT CAPACITY###
variable variance equal f_aves[8]-f_aves[7]*f_aves[7]
variable CV equal ${atoms}*${atoms}*${variance}/(f_aves[2]*f_aves[2])
''<u>>The equation for heat capacity is defined here, ${atoms} refer to the total number of atoms that the system possesses, ie. N. First, the equation for variance is defined, and then variable CV represents the final heat capacity value. </u>''
print "Averages"
print "--------"
print "Density: ${avedens}"
print "Stderr: ${errdens}"
print "Temperature: ${avetemp}"
print "Stderr: ${errtemp}"
print "Pressure: ${avepress}"
print "Stderr: ${errpress}"
print "HeatCapacity: ${CV}"
Figure 15. The Heat Capacity Plots with respect to Density = 0.2, 0.8 at varying temperatures.
For both densities, 0.2 and 0.8, the heat capacity is shown to decrease with increasing temperature, following close linear trends. Given that the heat capacity measures the ease in promoting the system to higher energy state, the property is related to Boltzmann distribution. And at higher temperatures, it is expected that there is greater uniformity among all energetic states and as a result, the states become increasingly saturated. This can be attributed to Schottky anomaly, which describes that the usual trend of increasing heat capacity with increasing temperature peaks at a certain temperature and subsequently decays due to saturated populations at higher levels. This is applicable for systems with finite number of energy levels. With regards to densities, the heat capacity is shown to possess a proportional relationship. which can be rationalized using the equation shown above. The heat capacity is an extensive property and therefore depends on the number of atoms in the systems; with increased number of atoms in the lattice, there is an expected increase in thermal diffusivity as the lattice can absorb more heat (having higher number of energy levels at which the system can be promoted to). However, given that the simulations utilize Newtonian laws and are by definition, not quantum systems, the absorption of energies involve moving to higher modes.
Structural Properties and the Radial Distribution Function
A radial distribution function describes the variation of density with distance from a reference point in a system of particles (in this case, the cubic lattice defined in the simulation input script). All of the g(r) calculations between the phases are normalized by dividing g(r) with the initial density of the lattice at every radial extension. The radial distribution functions connect macroscopic thermodynamic properties with the intermolecular interactions within fluids. As before, the system of atoms is described with a lattice-model of spherical particles, each interacting with one another using the 12-6 pair potential. Subjecting this lattice to different density and temperature external conditions result in the formation of the vapor, liquid and solid phases. The determination of such conditions are described by a Lennard-Jones phase diagram[4]. Experimental conditions are specified below:
Lennard-Jones Fluid Phase
Density
Temperature
(reduced unit)
Solid
1.18
0.5
Liquid
0.8
1.2
Gas
0.15
2.0
Slightly extreme conditions for each phase are chosen to prevent the occurrence of phase co-existence, which will distort the radial distribution function, g(r). Furthermore, the solid phase is simulated using the fcc lattice because it is necessary to allow the system to reach to its lowest energetic state first (for meaningful comparison between the radial distribution functions). This cannot be done for the solid phase in the simple cubic lattice, which is higher in energy than the fcc lattice, due to a smaller packing efficiency. The input scripts are used in the LAMMPS simulations, and the trajectory outputs of which are visualized using the VMD software. Radial distribution functions for all of the three phases are plotted below:
Figure 16. Comparison between the Radial Distribution Functions of Lennard-Jones Fluid in different phases (normalized).
The most evident difference between the three g(r) plots is the number of maxima for each phase: from solid to liquid to gas, there is a sharp decrease of maxima in radial distribution as the radial extension increases. Contrary to expectations that the radial distribution function is a steady decay with increasing radial extensions, all phases show a trend of oscillating peaks at shorter distances (the gas phase radial distribution function only has one peak).Due to the long range order of the Lennard-Jones interaction, the radial distribution function does not tend to zero at longer radial extensions, instead it tends to unity at 1. All g(r) functions tend to 0 for radial extensions smaller than 1, as this corresponds to unrealistic placement of atoms that results in repulsion (ie. no neighbouring particles can be found at these distances).
The solid g(r) has a more regular spacing between two interacting particles. And given that the radial distribution function also describes the probability of finding another atom/particle some distance away from a reference point, the function has a periodic series of maxima representing the high certainty that there are atoms across the radial extension.[5] The three maxima peaks in the solid phases on the radial extension versus g(r) plots are:
(1.075, 5.975)
(1.525, 1.223)
(1.825, 3.689)
Looking at the fcc unit cell, shown below, it is possible to allocate the radial distribution to each of the point above based on the radial extensions:
Figure 17. Labelled fcc unit cell showing three types of coordination with respect to a reference point.
Therefore, the lattice spacing described in the fcc cubic lattice corresponds to 1.525. The coordination number for each of the peaks can be visualized using expanding the periodic lattice structure. Using the corner lattice point as the reference point for this g(r) exercise, there are 12 coordination corresponding to the radial extension at 1.075. Similarly, by expanding the number of adjacent unit cells, there are 6 coordination that correspond to the lattice spacing, 1.525. Finally, the last peak has a higher radial distribution function than the second peak, suggesting a higher coordination number. At the third peak, there are 24 coordination with respect to the same reference point used earlier.[6]
In the liquid phase, there are slight irregular spacings such that the oscillatory pattern of the g(r) maxima is less pronounced compared to the plot from the solid phase. Short-range order Lennard-Jones potential predominates, but due to the increased disorder and irregularity between spacing, the disappearance of long-range order interaction is expected. This allows liquid g(r) to tend to unity a lot faster than the solid phase. It is also important to note the oscillatory behavior at short distances, which are derived from the particles interacting in the minimum of the Lennard-Jones potential well. This behaviour is expected given that the packing of atoms is similar to the solid phase, but the molecules have less restrictions on their movement.
Finally, the vapour radial distribution function has only one peak corresponding to the short-range order involved in Lennard-Jones interaction. At distances beyond that radial extension, the radial distribution function tends to unity after one peak, which suggests that the long range order of the Lennard Jones potential is at a even smaller magnitude compared to the liquid phase. Increased temperature and decreased density in the simulation of the vapour phase have resulted in higher irregularity within the lattice. The resulting g(r) trend is expected because by definition, the radial distribution function implicitly measures the density of the atoms surrounding the chosen reference point at any given distances.
Dynamic Properties and the Diffusion Coefficient
The following exercise seeks to describe the degree at which atoms in the system move/diffuse over the course of the simulation. A quantitative parameter that describes this is the diffusion coefficient, which can be calculated using the mean squared displacement (assuming a free random walk across the lattice) or the velocity autocorrelation function.
Mean Squared Displacement
The MSD measures the deviation over time in the particle's position over time with respect to a reference point. MSD can be modeled using the idea of a randomized 'walk' across the lattice, with no restriction on the directions. Based on the physical phase of the system at the given time, it can be expected that the diffusion of the atoms change dramatically: in the solid phase, the atoms are less able to diffuse due to the locking of their positions to maintain the physical integrity of the lattice; in the liquid and gaseous phases, the atoms are expected to move accordingly to Brownian motion and has less restrictions in terms of fixed atomic positions. As a consequence, the mean-squared displacement quickly saturates to a given value that corresponds to vibration motions. As shown at the plot below, this leads to a dramatic increase in the displacement once the simulation begins and plateau subsequently. Furthermore, the solid phase displays an oscillatory-type behaviour in its mean squared displacement, which further suggests that the atoms' displacements are behaving similarly to a damped harmonic oscillator. Solid state lattice can display a variety of diffusion mechanisms if impurities or defects are present. This can take the form of interstitial displacement, where an atom pushes one of the neighbours from its starting lattice site into another interstitial position.[7] However, given that the input scripts have only specified 'type 1' atoms with regular spacings, as shown below, this mechanism is not expected at all during any point of the simulation.
region box block 0 20 0 20 0 20
mass 1 1.0
On the other hand, the liquid and gaseous phases show close linearity in the mean squared displacement over time (suggesting that the cumulative distance traveled in the random walk process has consistent increase with time). The mean-squared displacement for the liquid phase achieves complete linearity, with R2 = 0.999, such that the trajectories and velocities of the atoms are completely randomized through frequent collisions in the system.
It is noteworthy to point out that the slope of the MSD in gaseous phase is ~10 times larger than that of the MSD in the liquid phase; this agrees with the fact that thermal motion is more pronounced in the gaseous phase and the Brownian motion becomes more apparent.Furthermore, the environment at which the atoms diffuse in the gaseous phase is fundamentally different from that of the liquid phase, given the lower density, which reduces the frequency of collision and therefore the atoms are expected to travel with constant velocity until a ballistic collision with other atoms occur. Until the atoms collide with other atoms in the gaseous phase, the atom moves with constant velocity and hence, the MSD is proportional to t2 , an exponential behaviour.
Small Scale: Mean Squared Displacement in Solid Phase
(with units of time steps)
Small Scale: Mean Squared Displacement in Liquid Phase
(with units of time steps)
Small Scale: Mean Squared Displacement in Gaseous Phase
(with units of time steps)
The simulations are repeated using the input files specifying a larger system, with one million atom simulations.
Large Scale: Mean Squared Displacement in Solid Phase
(with units of time steps)
Large Scale: Mean Squared Displacement in Liquid Phase
(with units of time steps)
Large Scale: Mean Squared Displacement in Gaseous Phase
(with units of time steps)
The diffusion coefficient is solved using the equation shown below and then converting number of steps to time (seconds), by dividing the value with the timestep (0.002).
Figure 18. Relationship between the diffusion coefficient and MSD
Diffusion Coefficient
(m2/s)
Small-Scale
Large-Scale
Percentage Difference
Solid Phase
8.33x10-7
4.167x10-6
80%
Liquid Phase
0.0833
0.0833
0%
Gaseous Phase
1.717
2.542
32%
It is noteworthy that the sample size does not change the phase behaviour with regards to the diffusive behaviors: the solid phase still display a plateau signifying the constant vibrational motion that atoms can only access due to strong interatomic forces keeping atoms in place, the liquid phase displays consistent Brownian motion and randomized velocities due to collisions, and the gaseous phase illustrating parabolic behaviour due to the system being more diluted. By definition of molecular dynamics simulations, larger sample sizes lead to a decrease in fluctuations, as the variance of a thermodynamic quantity is the inverse of N, the number of atoms. This is observed explicitly for the MSD plot for the solid phase, where the larger scale MSD contains reduced fluctuations in the plateau regime and initially during the simulation.
Velocity Autocorrelation Function (VACF)
Like the mean squared displacement, the velocity autocorrelation function describes the diffusive properties of a system of atoms. In general, transport coefficients such as the diffusion coefficient can be described through integrating time correlation functions. As described below, the autocorrelation function is such that the time origin can be shifted to any time in the simulation without affecting the overall function.
This is done in terms of velocities, where the function compares the velocity of the atom at time t to its velocity at time t + τ, as shown below. In this section, the input scripts are subjected to 5000 time-steps, with each timestep equal to 0.002. In order to simulate the different phases of the Lennard-Jones fluid in the system, the density and temperature parameters are changed accordingly, following the Lennard-Jones fluid phase diagram.
Figure 19. The velocity autocorrelation function
Given that the velocity function depends on the function for positions, the 1D harmonic oscillator also has an oscillatory VACF; the derivation of which is shown below:
Given that the sine function is an odd function and that the 'positive' areas under its curve equal to the 'negative' areas in the equation shown above, extension to infinity involves cancellation of both net 'areas', leaving to an integral equal to zero. This leaves the VACF solution for the 1D harmonic oscillator to be equal to cos(ωτ).
Figure 20. Comparison between the harmonic oscillator VCAF with solid and liquid phases of LJ fluid.
Small Scale: VACF in Small-Scale Simulation of Solid Phase
(with units of time steps)
Small Scale: VACF in Small-Scale Simulation of Liquid Phase
(with units of time steps)
Small Scale: VACF in Small-Scale Simulation of Gaseous Phase
(with units of time steps)
Gallery 3: The VACF over all simulated time steps for small-scale systems of Lennard-Jones fluid
Large Scale: VACF in Small-Scale Simulation of Solid Phase
(with units of time steps)
Large Scale: VACF in Small-Scale Simulation of Liquid Phase
(with units of time steps)
Large Scale: VACF in Small-Scale Simulation of Gaseous Phase
(with units of time steps)
Gallery 4: The VACF over all simulated time steps for large-scale systems of Lennard-Jones fluid, including 1 million atoms.
Comparisons with the 1D Harmonic Oscillator's VACF
A key feature of the 1D harmonic oscillator's VACF is the oscillatory behaviour along with the lack of decay across the time steps in the simulation. this can be understood, because, by definition of a harmonic oscillator, the restoring force is constant and that the system is in an idealized case. However, this is not the case for the Lennard-Jones fluid, given that there are stronger interatomic forces due to closer packing in a more dense environment. In the solid phase, the atoms have fixed regular positions and as a result can be assumed to have a damped harmonic oscillatory behaviour. As the atoms vibrate back and forth, but cannot escape from their original positions in a fixed lattice, the VACF oscillates. Negative correlation implies that the particle is moving in the opposite direction, and hence implies a damped harmonic oscillation is in progress. Similarly, the liquid phase of the Lennard Jones fluid experiences strong interatomic forces and a relatively high density in the system. However, due to the inclusion of Brownian motion, there is a higher rate of diffusion on the same time scale, which explains why the VACF plot is oscillatory at the beginning but decay rapidly relative to the solid phase. The damping process for the liquid phase of VACF is the result of more frequent collisions between two atoms, which produces different velocities as a consequence.
All VACF plots show a steady decay to zero, as the particles' velocities decorrelate with time due to increasingly diffusive behaviour. Unlike in the case of the solid, the atoms in the liquid phase does not observe high long-range interactions due to a less-dense environment and therefore diffusive properties become more dominant, resulting in a faster decay to zero correlation.
Calculating the Diffusion Coefficients
The diffusion coefficient is related to the velocity autocorrelation function through the following integral:
Figure 21. Relationship between the diffusion coefficient and velocity autocorrelation function
By performing the trapezium rule across the entire simulation's time-steps, the area under the VACF curve can be approximated. By definition, this would correspond to the total displacement of the system from the initial point of the simulation to the end. The running integral is calculated by finding the area under the curve between each timestep (Δt =1 ) for that step and then adding it to the total cumulative area summed before this current time-step. As a result, the total integral can be obtained from the last data point of the running integral plot and subsequently converting the time-step to time in units. This methodology results in the following running integral plots:
Small Scale: Running Integral of VACF in Solid Phase
(with units of time steps)
Small Scale: Running Integral of VACF in Liquid Phase
(with units of time steps)
Small Scale: Running Integral of VACF in Gaseous Phase
(with units of time steps)
Gallery 5: The running integrals of VACF of the small-scale simulations of Lennard-Jones fluid in three different physical phases.
Large Scale: Running Integral of VACF in Solid Phase
(with units of time steps)
Large Scale: Running Integral of VACF in Liquid Phase
(with units of time steps)
Large Scale: Running Integral of VACF in Gaseous Phase
(with units of time steps)
Gallery 6: The running integrals of VACF of the large-scale simulations (1 million atoms) of Lennard-Jones fluid in three different physical phases.
Diffusion Coefficients
(m2/s)
MSD
(Small-Scale Simulations)
MSD
(Large-Scale Simulations)
VACF Integral
(Small-Scale Simulations)
VACF Integral
(Large-Scale Simulations)
Solid Phase
8.33x10-7
4.167x10-6
5.285x10-5
4.553x10-5
Liquid Phase
0.0833
0.0833
0.122
0.113
Gaseous Phase
1.717
2.542
1.808
4.0855
Table: Summary of Diffusion Coefficient calculations using both MSD and VACF methods
As in the MSD's case, the switch to a larger sample size does not produce significantly different results for diffusion coefficients, given that both coefficients are still in the same order of magnitude. Any differences in the outputs would originate from how the initial densities and temperatures are defined for the smaller-scale simulations, which would impact the extent of Brownian motion and frequency of ballistic collisions, and therefore the overall diffusivity of the system. It should also be noted from the VACF curves shown in galleries 4 and 5, the velocity autocorrelation function can dip into the negative values. This result from the fact that the atoms can move in the opposite velocities during any given time-steps (the random walk process does not restrict atoms from moving in opposite directions, which again justified why MSD needs to square all the velocities). The running integral plots all display a plateau-like behavior after high number of time-steps. this indicates that the change in the area under VCAF curve becomes increasingly negligible, which is expected as the system tends to maximum diffusivity.
The most evident error involved in the VACF method for calculation is the overestimation of the trapezium area under the curve. This results from using a larger difference in time-step for calculating the areas. Furthermore, this error is expected to be propagated given that each area is added cumulatively to give the total integral. On the other hand, if the VACF curve is as uniform as in the case of the 1D harmonic oscillator, evaluating the integral would not require the use of trapezium rule and therefore this error would not be associated with the calculations. The VACF calculation is another example why the choice of time-step is important in mitigating errors, as shown earlier in previous exercises. Choosing a smaller time-step to derive the diffusion coefficient would be computationally demanding, and hence any improvements to the simulations should examine the most appropriate time-step that matches the evolution of the lattice. A topical research topic in this area could investigate the use of multiple time-steps to capture ranges of processes that change with different time scales, instead of using a one-size-fits-all time step to evaluate multiple thermodynamic properties.
In comparing the MSD methodology with VAC, the two functions are related through integrating the velocity over a chosen interval of time. Of the three phases observed, the liquid phase has the highest reliability given the absolute linearity observed in MSD plot (which indicates that the trendline's slope is accurate to be used in the diffusion coefficient) and that the VACF plot is reasonably uniform, such that using trapezium rule would not incur as much error.
Conclusion
In this experiment, a MD simulation of a Lennard-Jones fluid is performed using Newtonian laws, placing the system inside a simulation box that abides by periodic boundary conditions and applying ensemble averages in calculating the thermodynamic properties of the system. This is achieved by using classical particle approximation due to difficulties in using quantum mechanics to describe a N-body system accurately. Analysis and understanding of the Velocity Verlet Algorithm indicates that there are inherent errors involved in numerical integration, which arises from large time-steps and excluding terms from the expansion of positions and velocities.
The Lennard-Jones fluid is then simulated in a defined ensemble with different time-steps (canonical ensemble) and then its thermodynamics properties are measured. By observing how closely the system follows the law of energy conservation at equilibration, a suitable time-step is then defined (in this case, 0.0025). It is also observed that time-steps that are too high result in an 'explosion' in the lattice, where highly unfavourable repulsion take place due to atomic collisions. By changing the ensemble to the isobaric-isothermal ensemble, the equation of state for Lennard-Jones fluid can be derived. This is tested using two different densities and five different initial temperatures. The results show large discrepancies from the ideal gas law, which only increase with higher densities and lower temperature conditions. This is rationalized by the inclusion of strong short-range and long-range interactions between the atoms, and hence kinetic energy does not entirely describe the system, as it would in an idealized case. Thus, high thermal energy is required to allow the system to converge with the ideal gas law.
Any thermodynamic quantities' fluctuations are described by the heat capacity of the system, which depends on the sample size and the variance in energy. The resulting trend is not as expected (which is that increasing temperature increases the heat capacity). A viable explanation is that the system has reached a saturation in terms of populating available modes or that the energy spacing between modes is relatively small such that little heat is required to promote the system into higher excited levels.
The radial distribution functions are calculated using the VMD software for three different phases of Lennard Jones fluid. For the solid phase, it was shown that using the simple cubic starting lattice does not exhibit accurate radial distributions due to the fact that a fcc starting lattice has a lower ground-state energy. Furthermore, the continuous oscillation in the radial distribution function suggests that the solid phase has a high degree of long-range interactions, and using the first three peaks on the g(r) curves, the lattice structure can be derived. The liquid phase displays minimal long-range interactions given that the atom spacing in the liquid phase mirrors that of the solid phase but due to smaller density, longer-range interactions become increasingly negligible.
Finally, the system's diffusivity was determined using mean-squared-displacement and the VACF method. MSD models a random walk process where a starting atom can 'wander' across the lattice, and the sum of its displacements would quantify the diffusivity (or the ease at which diffusion can occur in the system). Vibration motions are characterized in the case of the solid, which shows a plateau region as atoms could only oscillate while locked in place by strong interactions. Collisions become more frequent in the liquid phase and thus, velocities are randomized throughout the simulation. A less dense gaseous phase would have reduced frequency of collisions with other atoms and have velocities of a 'freely travelling' atom, and hence display a quadratic trend at the beginning due to proportionality to t2. As collisions become more frequent over time due to attractions between the atoms, the MSD observes in the gas phase tends to linearity. The VACF methods support this observation and show that attractive interactions present (by nature of a Lennard-Jones fluid) would result in a decorrelation of velocities over time due to diffusivity and damping behaviour.
Finally, as discussed in their respective sections, the accuracy of molecular dynamics simulations can be tested by altering some of the assumptions made during the calculations. Further investigations could involve altering the cutoff distance for the Lennard-Jones potential, which can be tailored to the phase diagram of the Lennard-Jones fluid. In addition, investigating the use of multiple time-steps specific to different thermodynamic quantities parallel to each other during one simulation is a logical progression to improve MD calculations, as many numerical integration methods depend on the time intervals at which data is collected.
References
↑Sousa N, J.J Saenz, F. Scheffold, A. Garcia-Martin and L.S Froufe-Perez, Self-diffusion and structural properties of confined fluids in dynamic coexistence, 2016.















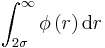
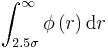
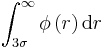



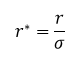
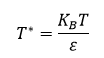
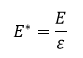






.PNG)
.PNG)
















.PNG)
2.PNG)






























