
Rep:Mod:JT3818:2019 1
--Jt3818
Part 1
Summary information
File:JT3818 NH3 OPTF POP 2 DISTANCE.LOG
| Molecule name | ammonia, azane |
|---|---|
| Calculation method | RB3LYP |
| Basis set | 6-31G(d,p) |
| Final energy | –56.55776873 a.u. |
| Point group | C3v |
Item Value Threshold Converged? Maximum Force 0.000004 0.000450 YES RMS Force 0.000004 0.000300 YES Maximum Displacement 0.000072 0.001800 YES RMS Displacement 0.000035 0.001200 YES Predicted change in Energy=-5.986278D-10 Optimization completed.
Structure
Optimized interactive 3D structure of ammonia |
Calculated bond length is 1.02 Å, and the bonding angle is 106°. These values are in accordance with experimental data (bond length 101.7 pm and 107.8° [1]).
Part 2
Vibration analysis
| wavenumber [cm–1] | symmetry | intensity | ||
|---|---|---|---|---|
| 1 | 
|
1089 | A1 | 145 |
| 2 | 
|
1694 | E | 14 |
| 3 | 
|
1694 | E | 14 |
| 4 | 
|
3461 | A1 | 1 |
| 5 | 
|
3590 | E | 0 |
| 6 | 
|
3590 | E | 0 |
Questions & Answers
How many modes do you expect from the 3N-6 rule? 6 vibration modes were expected and were calculated.
Which modes are degenerate (ie have the same energy)? Modes 2 and 3 (1694 cm–1), and 5 and 6 (3590 cm–1) are degenerate.
Which modes are "bending" vibrations and which are "bond stretch" vibrations? bending: 1, 2, 3 stretching: 4, 5, 6
Which mode is highly symmetric? highly symmetric: 4
One mode is known as the "umbrella" mode, which one is this? umbrella: 1
How many bands would you expect to see in an experimental spectrum of gaseous ammonia? 2 band (1-3 active, but 2+3 degenerate and 4-6 inactive).
Atomic charges
Partial atomic charges are N: –1.125, H: 0.375. The nitrogen atom is bearing the negative charge since it is more electronegative than hydrogen.
Part 3
Nitrogen
Summary information
| Molecule name | dinitrogen |
|---|---|
| Calculation method | RB3LYP |
| Basis set | 6-31G(d,p) |
| Final energy | –109.52412868 a.u. |
| Point group | D∞h |
Item Value Threshold Converged?
Maximum Force 0.000001 0.000450 YES
RMS Force 0.000001 0.000300 YES
Maximum Displacement 0.000000 0.001800 YES
RMS Displacement 0.000000 0.001200 YES
Predicted change in Energy=-2.936213D-13
Optimization completed.
Structure
Optimized interactive 3D structure of dinitrogen |
Calculated bond length is 1.11 Å (literature value: 1.098 Å [2]).
Vibration analysis
Since dinitrogen is a linear diatomic molecule, one vibration mode is expected (3N–5, where N=2). As can be seen in the table below, vibration is symmetric and does not involve change so it is IR inactive.
Harmonic frequencies (cm**-1), IR intensities (KM/Mole), Raman scattering
activities (A**4/AMU), depolarization ratios for plane and unpolarized
incident light, reduced masses (AMU), force constants (mDyne/A),
and normal coordinates:
1
SGG
Frequencies -- 2457.3342
Red. masses -- 14.0031
Frc consts -- 49.8198
IR Inten -- 0.0000
| wavenumber [cm–1] | symmetry | intensity | ||
|---|---|---|---|---|
| 1 | 
|
2457 | SGG | 0 |
Charges
Dinitrogen is a homonuclear diatomic molecule. Therefore, zero partial charges can be expected which was also the result of the calculation.
Hydrogen
Summary information
| Molecule name | dihydrogen |
|---|---|
| Calculation method | RB3LYP |
| Basis set | 6-31G(d,p) |
| Final energy | –1.17853936 a.u. |
| Point group | D∞h |
Item Value Threshold Converged? Maximum Force 0.000000 0.000450 YES RMS Force 0.000000 0.000300 YES Maximum Displacement 0.000000 0.001800 YES RMS Displacement 0.000001 0.001200 YES Predicted change in Energy=-1.164080D-13 Optimization completed.
Structure
Optimized interactive 3D structure of dihydrogen |
Bond length is calculated to be 0.743 Å which is in excellent match with the literature value 0.74 Å [3].
Vibration analysis
Dihydrogen is a linear diatomic molecule; one vibration mode is expected (3N–5, where N=2). As can be seen in the table below, vibration is symmetric and does not involve a change in dipole moment, so it is IR inactive.
Harmonic frequencies (cm**-1), IR intensities (KM/Mole), Raman scattering
activities (A**4/AMU), depolarization ratios for plane and unpolarized
incident light, reduced masses (AMU), force constants (mDyne/A),
and normal coordinates:
1
SGG
Frequencies -- 4465.6824
Red. masses -- 1.0078
Frc consts -- 11.8416
IR Inten -- 0.0000
| wavenumber [cm–1] | symmetry | intensity | ||
|---|---|---|---|---|
| 1 | 
|
4466 | SGG | 0 |
Charges
As it is with dinitrogen molecule, dihydrogen has equal electron distribution, so each atom is bearing a zero partial charge.
Comparison of the hydrogen bond length with crystallographic data
Calculated hydrogen bond was compared with the crystal structures of transition metal complexes having coordinated molecular hydrogen. In total, 73 compounds were found in the CCDC database (complexes with more than one transition metal were ignored). The variation of H—H bond length can be seen in the histogram below. The most frequent bond length was approximately 0.8 Å (16 hits). An example of the complex with such bond length is EWUQIW; its crystal structure is shown below. It is ruthenium complex and the H—H bond length is 0.816 Å which is longer than the calculated value (0.743 Å). This elongation can be possibly explained by weakening the H—H by the interactions of metal d-orbitals and σ* antibonding dihydrogen orbitals. [4] CCDC reference: [5]
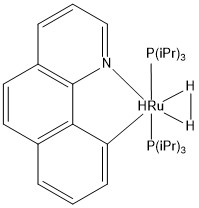
|
| Hydrido-(dihydrogen-H,H')-bis(tri-isopropylphosphine)-(benzo(h)quinolin-10-yl-C,N)-ruthenium |

|
| Crystal structure of EWUQIW complex. |

|
| Histogram showing the H—H bond length distribution, x-axis is in Å units. |
Reaction energy of ammonia formation
Calculation of the reaction energy change:
E(NH3)= –56.55776873 a.u.
2*E(NH3)= –113.11553746 a.u.
E(N2)= –109.52412868 a.u.
E(H2)= –1.17853936 a.u
3*E(H2)= –3.53561808 a.u.
ΔE=2*E(NH3)-[E(N2)+3*E(H2)]= (–113.11553746 – [–109.52412868 –3.53561808]) a.u. = -0.0557907 a.u. ≈ –146.5 kJ/mol
The calculated value of energy change is approximately equal to –146.5 kJ/mol, so the product (ammonia) is more stable than the reactants (nitrogen and hydrogen) since the energy change is negative.
Part 4+5
For this part of the computational lab, I chose PF3Cl2 as a molecule for investigation. I firstly fought, that the structure would look like that the two chlorine atoms were in axial positions while the three fluorine atoms would be in equatorial positions. However, after calculating the energies of the possible isomers, the apparently more stable structure was found (unfortunately, I found this out after having had already written the subsection below). Therefore, the isomerism of phosphorus trifluoride dichloride was researched in literature. In the subsections below, the computational analysis of possible isomers and the experimental evidence from the literature are provided.
Phosphorus Trifluoridedichloride – Chlorine Atoms Axial
The structure of PF3Cl2 with two chlorine atoms (D3h symmetry point group) was erroneously assigned by Brockway and Beach using a diffraction experiment. [6] Moreover, this wrong information can be also found in various discussion forums (e.g. [7]).
Summary information
| Molecule name | Phosphorus Trifluoride Dichloride |
|---|---|
| Calculation method | RB3LYP |
| Basis set | 6-31G(d,p) |
| Final energy | –1561.34405351 a.u. |
| Point group | D3h |
Item Value Threshold Converged? Maximum Force 0.000008 0.000450 YES RMS Force 0.000003 0.000300 YES Maximum Displacement 0.000046 0.001800 YES RMS Displacement 0.000014 0.001200 YES Predicted change in Energy=-3.841754D-10 Optimization completed.
Structure
Optimized interactive 3D structure of phosphorus trifluoride dichloride |
Calculated bond lengths are P—F 1.58 Å and P—Cl 2.10 Å. Bonding angles are the same as it would be expected by VSEPR theory: F—P—F 120° and F—P—Cl 90°. Since chlorine atoms are larger (atomic radius increases down the group), chlorine atoms are found in axial positions, and fluorine atoms are in the equatorial positions. Thanks to this arrangement, chlorine atoms are as far apart from each other as possible.
Vibration analysis
Phosphorus trifluoride dichloride is a nonlinear polyatomic molecule, so 12 vibration modes are expected (3N–6, where N=6). Each vibration mode, calculated wavenumber, symmetry and intensity is shown in the table below.
Harmonic frequencies (cm**-1), IR intensities (KM/Mole), Raman scattering
activities (A**4/AMU), depolarization ratios for plane and unpolarized
incident light, reduced masses (AMU), force constants (mDyne/A),
and normal coordinates:
1 2 3
E' E' ?A
Frequencies -- 116.6886 116.6893 348.4439
Red. masses -- 23.1661 23.1661 23.2546
Frc consts -- 0.1858 0.1859 1.6635
IR Inten -- 0.0666 0.0666 12.8683
4 5 6
?A ?A E"
Frequencies -- 348.4440 352.8630 370.8283
Red. masses -- 23.2546 34.5184 20.7761
Frc consts -- 1.6635 2.5323 1.6833
IR Inten -- 12.8699 0.0000 0.0000
7 8 9
E" A2" A2"
Frequencies -- 370.8284 450.7731 588.5484
Red. masses -- 20.7761 24.3312 29.0515
Frc consts -- 1.6833 2.9129 5.9290
IR Inten -- 0.0000 2.0732 682.8687
10 11 12
A1' E' E'
Frequencies -- 707.1356 984.6328 984.6359
Red. masses -- 19.1341 23.8162 23.8162
Frc consts -- 5.6372 13.6041 13.6042
IR Inten -- 0.0000 188.8169 188.8102
| wavenumber [cm–1] | symmetry | intensity | ||
|---|---|---|---|---|
| 1 | 
|
117 | E' | 0 |
| 2 | 
|
117 | E' | 0 |
| 3 | 
|
349 | ?A | 13 |
| 4 | 
|
349 | ?A | 13 |
| 5 | 
|
352 | ?A | 0 |
| 6 | 
|
371 | E" | 0 |
| 7 | 
|
371 | E" | 0 |
| 8 | 
|
451 | A2" | 2 |
| 9 | 
|
451 | A2" | 2 |
| 10 | 
|
707 | A1' | 0 |
| 11 | 
|
985 | E2' | 189 |
| 12 | 
|
985 | E' | 189 |
As can be seen from the computed wavenumbers, 5 pairs of 12 vibration modes are degenerate. Some of the vibration modes are also inactive. Therefore only 3 bands would be expected in the IR spectrum.
Charges
Both chlorine and fluorine have a higher electronegativity than central phosphorus atom, so they are bearing partial negative charges while the phosphorus has a partial positive charge. Computed partial charges can be seen on the figure below. Since fluorine is more electronegative than chlorine, fluorine atoms bear more negative charges than chlorine atoms. As the molecule has symmetrical charge distribution, the dipole moment is equal to zero.

|
| Distribution of partial charges in PF3Cl2 molecule |
Phosphorus Trifluoride Dichloride – Chlorine Atoms Equatorial
Summary information
| Molecule name | Phosphorus Trifluoride Dichloride |
|---|---|
| Calculation method | RB3LYP |
| Basis set | 6-31G(d,p) |
| Final energy | –1561.34405351 a.u. |
| Point group | C2v |
Item Value Threshold Converged?
Maximum Force 0.000032 0.000450 YES
RMS Force 0.000011 0.000300 YES
Maximum Displacement 0.000122 0.001800 YES
RMS Displacement 0.000054 0.001200 YES
Predicted change in Energy=-6.037701D-09
Optimization completed.
Structure
Optimized interactive 3D structure of phosphorus trifluoride dichloride |
Calculated bond lengths are: P—Feq 1.58 Å, P—Fax 1.61 Å and P—Cl 2.04 Å. Literature bond distances(P—Fax 1.59 Å and P—Cl 2.04 Å) are in very good match with calculated values.[8] Bonding angles are slightly different than it expected by VSEPR theory for trigonal bipyramidal structure: Cl—P—Cl 122.3° and Fax—P—Cl 90.3°. As chlorine atoms are larger, the Cl—P—Cl is larger than 120°.
Vibration analysis
Same as with its isomer, 12 vibration modes are expected (3N–6, where N=6). Each vibration mode, calculated wavenumber, symmetry and intensity is shown in the table below.
Harmonic frequencies (cm**-1), IR intensities (KM/Mole), Raman scattering
activities (A**4/AMU), depolarization ratios for plane and unpolarized
incident light, reduced masses (AMU), force constants (mDyne/A),
and normal coordinates:
1 2 3
A1 B2 B1
Frequencies -- 112.3152 153.6172 325.1533
Red. masses -- 28.1268 20.2923 24.6594
Frc consts -- 0.2090 0.2821 1.5361
IR Inten -- 0.0385 0.4686 3.8150
4 5 6
A2 A1 B2
Frequencies -- 343.2104 389.2581 407.6122
Red. masses -- 22.0819 28.2684 24.2031
Frc consts -- 1.5325 2.5236 2.3693
7 8 9
A1 B1 B2
Frequencies -- 473.9946 486.2914 641.7258
Red. masses -- 23.0787 19.5878 30.5362
Frc consts -- 3.0550 2.7292 7.4091
IR Inten -- 47.7328 11.8326 375.7565
10 11 12
A1 A1 B1
Frequencies -- 676.9253 913.4476 951.8889
Red. masses -- 19.3030 23.0808 24.2328
Frc consts -- 5.2114 11.3467 12.9368
IR Inten -- 0.0433 198.1455 280.1533
| wavenumber [cm–1] | symmetry | intensity | ||
|---|---|---|---|---|
| 1 | 
|
112 | A1 | 0 |
| 2 | 
|
154 | B2 | 0(0.47) |
| 3 | 
|
325 | B1 | 4 |
| 4 | 
|
343 | A2 | 2 |
| 5 | 
|
389 | A1 | 3 |
| 6 | 
|
408 | B2 | 2 |
| 7 | 
|
473 | A1 | 2 |
| 8 | 
|
486 | B1 | 12 |
| 9 | 
|
642 | B2 | 378 |
| 10 | 
|
677 | A1 | 0 |
| 11 | 
|
913 | A1 | 198 |
| 12 | 
|
952 | B1 | 280 |
Judging by the calculated IR intensities and frequencies, none of the modes is degenerate, and three of the 12 modes are expected to be IR inactive.
Charges
The order of electronegativities is F>Cl>P, which is reflected on the partial charges (phosphorus atom has a positive charge, while the fluorine negative, value of negative charge on chlorine is smaller than on fluorine atom). Unlike its isomer with axial chlorine atoms, PF3Cl2 with chlorine atoms in equatorial position has an uneven charge distribution. Therefore, it has a dipole moment, and its direction is shown below with the values of partial charges.

|
| Distribution of partial charges in PF3Cl2 molecule |
Phosphorus trifluoride dichloride – one chlorine atom equatorial, one axial
To prevent this assignment from being too long, less information about the isomer of PF3Cl2 with one chlorine atom in axial and one in equatorial position is provided than for the previous ones. Information about vibration modes, bond lengths, angles and charges can be accessed from the LOG file below using Gaussview software. File:JT3818 PF3CL2 AX+EQ.LOG
Summary information
| Molecule name | Phosphorus Trifluoride Dichloride |
|---|---|
| Calculation method | RB3LYP |
| Basis set | 6-31G(d,p) |
| Final energy | –1561.34848132 a.u. |
| Point group | Cs |
Item Value Threshold Converged? Maximum Force 0.000084 0.000450 YES RMS Force 0.000031 0.000300 YES Maximum Displacement 0.000619 0.001800 YES RMS Displacement 0.000261 0.001200 YES Predicted change in Energy=-7.025217D-08 Optimization completed.
Comparison of isomers
As can be seen in the table below, there are subtle differences in the energy (on the second decimal place) of the three possible isomers of PF3Cl2. The most stable isomer is expected to be the one with both chlorine atoms in equatorial positions. This is in accordance with the experimental data. [8] [9] For example, it is possible to see two different signals in 19F NMR at –124 °C which directly rules out the originally reported structure [6] with two chlorine atoms in axial positions. Also, the molecule possesses a dipole moment which PF3Cl2 with axial chlorine atoms does not have. [9] However, the isomer with one chlorine atom in axial and one in equatorial position would also have 2 different fluorine environments and a dipole moment. the evidence for the actual structure is the data obtained from IR, Raman and other advanced spectroscopy methods. [8][9]
| chlorine atoms axial | chlorine atoms equatorial | one axial, one equatorial | exp. evidence | |
|---|---|---|---|---|

|

|

|
||
| Energy | -1561.34405351 a.u. | -1561.35287071 a.u. | −1561.34848132 a.u. | |
| Point group | D3h | C2v | Cs | |
| Dipole moment | no | yes | yes | yes |
| Types of fluorine environments | 1 | 2 | 2 | pair of doublets |
Molecular orbitals (chlorine atoms equatorial)
PF3Cl2 has 76 electrons in total (15 from phosphorus atom, 9 from each fluorine atom and 17 from each chlorine atom). Therefore, 38 molecular orbitals are occupied. The lowest energy orbitals (not shown, the ones formed from 1s and 2s atomic orbitals) have the electron densities concentrated very close to the nucleus because of the high effective nuclear charges. Five representative molecular orbitals are presented in the tables below, the first one is occupied, and the following four are filled.

|

|
LUMO is formed as a destructive combination of 3s orbital from phosphorus atom, 2p orbitals from all fluorine atoms and 3p orbitals from chlorine atoms. Therefore, it is an antibonding orbital. It is high in energy (but its energy is still negative unlike LUMO+1 and higher MOs).

|

|
HOMO is formed by a destructive overlap of 2p orbitals from axial fluorine atoms and 3p orbitals from chlorine atoms. Therefore, it is an antibonding orbital. It is high in energy.

|

|
MO34 is formed as both constructive and destructive combination of 3p orbital from phosphorus atom, 2p orbitals from axial fluorine atoms and 3p orbitals from chlorine atoms. Therefore, it does not have a significant effect on bonding. It is relatively low in energy.

|

|
MO27 is formed as a constructive combination of 3p orbital from phosphorus atom, 2p orbitals from all fluorine atoms and 3p orbitals from chlorine atoms. Therefore, it is a bonding orbital. It is relatively low in energy.

|

|
MO24 is formed as a constructive combination of 3s orbital from phosphorus atom, 2p orbitals from all fluorine atoms and 3p orbitals from chlorine atoms. Therefore, it is a bonding orbital and should have a significant effect on bonding. It is low in energy.
References
- ↑ Greenwood, N. N.; Earnshaw, A. (1997) Chemistry of the Elements (2nd ed.), Oxford:Butterworth-Heinemann, pp. p. 423 ISBN: 0-7506-3365-4.
- ↑ Greenwood, N. N.; Earnshaw, A. (1997) Chemistry of the Elements (2nd ed.), Oxford:Butterworth-Heinemann, pp. p. 483 ISBN: 0-7506-3365-4.
- ↑ Atkins, P.; Overton, T.; Rourke, J.; et al. (2009) Inorganic Chemistry (5th ed.), OUP, pp. p. 58 ISBN: 978-0199236176.
- ↑ Toreki, R. (2015). The Organometallic HyperTextBook: Dihydrogen Complexes. [online] Ilpi.com. Available at: http://www.ilpi.com/organomet/dihydrogen.html [Accessed 7 Mar. 2019].
- ↑ https://www.ccdc.cam.ac.uk/structures/search?id=doi:10.5517/cc7pgv6&sid=DataCite
- ↑ 6.0 6.1 L. O. Brockway and J. Y. Beach, The Electron Diffraction Investigation of the Molecular Structures of (1) Phosphorus Oxytrichloride, Oxydichlorofluoride, Oxychlorodifluoride, Oxytrifluoride, Fluorodichloride, Pentafluoride, and Trifluorodichloride, and of (2) Disilane, Trichlorosilane, and Hexachlorodisilane, Journal of the American Chemical Society, 1938, 60, 1836–1846.
- ↑ https://uk.answers.yahoo.com/question/index?qid=20061228225256AAA1RbO&guccounter=1&guce_referrer=aHR0cHM6Ly93d3cuZ29vZ2xlLmNvbS8&guce_referrer_sig=AQAAAIpn9AKYzd9HUWAR9hKJxlmD5_OBV9X6TnO8149G2gyueH1iZM01ZATII-WkrhjOvCiEwbW7tYwdrBflfh_j1N93z00C4DjI35oLPMwDeinqSGJSq6guAz7xZnl2CWnXv3zcVPm1r6S23n285uBE9tSKyi04crs895eEKPj_nsLb)
- ↑ 8.0 8.1 8.2 . E. Griffiths, R. P. Carter Jr. and R. R. Holmes, Molecular Structures of PCl4F, PCl3F2, PCl2F3, and PF5: Infrared and Low‐Temperature Raman Vibrational Spectra, The Journal of Chemical Physics, 1964, 41, 863–876.
- ↑ 9.0 9.1 9.2 9.3 R. R. Holmes, R. P. Carter Jr. and G. E. Peterson, Molecular Structures of PCl4F, PCl3F2, and PCl2F3: Pure Chlorine Nuclear Quadrupole Resonance and Low Temperature F19Nuclear Magnetic Resonance Spectra, Inorganic Chemistry, 1964, 3, 1748–1754.
Marking
Note: All grades and comments are provisional and subjecct to change until your grades are officially returned via blackboard. Please do not contact anyone about anything to do with the marking of this lab until you have recieved your grade from blackboard.
Wiki structure and presentation 1/1
Is your wiki page clear and easy to follow, with consistent formatting?
YES
Do you effectively use tables, figures and subheadings to communicate your work?
YES
NH3 1/1
Have you completed the calculation and given a link to the file?
YES
Have you included summary and item tables in your wiki?
YES
Have you included a 3d jmol file or an image of the finished structure?
YES
Have you included the bond lengths and angles asked for?
YES
Have you included the “display vibrations” table?
YES
Have you added a table to your wiki listing the wavenumber and intensity of each vibration?
YES
Did you do the optional extra of adding images of the vibrations?
YES
Have you included answers to the questions about vibrations and charges in the lab script?
YES
N2 and H2 0.5/0.5
Have you completed the calculations and included all relevant information? (summary, item table, structural information, jmol image, vibrations and charges)
YES, you could have explained that the charges are 0 as the electronegativities are equal.
Crystal structure comparison 0.5/0.5
Have you included a link to a structure from the CCDC that includes a coordinated N2 or H2 molecule?
YES
Have you compared your optimised bond distance to the crystal structure bond distance?
YES
Haber-Bosch reaction energy calculation 1/1
Have you correctly calculated the energies asked for? ΔE=2*E(NH3)-[E(N2)+3*E(H2)]
YES
Have you reported your answers to the correct number of decimal places?
YES
Do your energies have the correct +/- sign?
YES
Have you answered the question, Identify which is more stable the gaseous reactants or the ammonia product?
YES
Your choice of small molecule 4.5/5
Have you completed the calculation and included all relevant information?
YES
Have you added information about MOs and charges on atoms?
You have done a good job of presenting this information, well done! You could have explained the charges using an electronegativity argument. It was asked to comment on the MOs occupancy, this is the only part you missed. Additionally you labelled MO 38 as anti-bonding even though no electron density is found on the bonds between two atoms. This is a non-bonding orbital.
Independence 1/1
If you have finished everything else and have spare time in the lab you could: Check one of your results against the literature, or Do an extra calculation on another small molecule, or
YES - well done!
Do some deeper analysis on your results so far