
Rep:Mod:JR2552
Jack Rogan - Module 2
Week 1 - Using Gaussian
BH3 Optimisation
B3LYP Basis set
BH3 was modelled, and optimised, first, with a B3LYP method and 3-21G basis set.
Result: JR_BH3_OPTIMISATION

| B-H Bond Distance / Å | 1.19 |
|---|---|
| H-B-H Bond angle / ° | 120.0 |
| File Type | .log |
|---|---|
| Calculation Type | FOPT |
| Calculation Method | RB3LYP |
| Basis Set | 3-21G |
| Final Energy / au | -26.462 |
| Gradient / au | 0.000 |
| Dipole Moment / D | 0.00 |
| Point Group | D3H |
| Calculation Time / min:sec | 1:54 |
Output
Item Value Threshold Converged?
Maximum Force 0.000413 0.000450 YES
RMS Force 0.000271 0.000300 YES
Maximum Displacement 0.001610 0.001800 YES
RMS Displacement 0.001054 0.001200 YES
Predicted change in Energy=-1.071764D-06
Optimization completed.
-- Stationary point found.
----------------------------
! Optimized Parameters !
! (Angstroms and Degrees) !
-------------------------- --------------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------------------
! R1 R(1,2) 1.1935 -DE/DX = 0.0004 !
! R2 R(1,3) 1.1935 -DE/DX = 0.0004 !
! R3 R(1,4) 1.1935 -DE/DX = 0.0004 !
! A1 A(2,1,3) 120.0 -DE/DX = 0.0 !
! A2 A(2,1,4) 120.0 -DE/DX = 0.0 !
! A3 A(3,1,4) 120.0 -DE/DX = 0.0 !
! D1 D(2,1,4,3) 180.0 -DE/DX = 0.0 !
--------------------------------------------------------------------------------
GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
6-31G(d,p) Basis set
The geometry was further optimised using the same method, but a more accurate - and calculation-intensive - 6-31G basis set instead.
Result: JR_BH3_OPTIMISATION_2

| B-H Bond Distance / Å | 1.19 |
|---|---|
| H-B-H Bond angle / ° | 120.0 |
| File Type | .log |
|---|---|
| Calculation Type | FOPT |
| Calculation Method | RB3LYP |
| Basis Set | 6-31G(d,p) |
| Final Energy / au | -26.615 |
| Gradient / au | 0.000 |
| Dipole Moment / D | 0.00 |
| Point Group | CS |
| Calculation Time / min:sec | 0:07 (On HPC) |
Output
Item Value Threshold Converged?
Maximum Force 0.000005 0.000450 YES
RMS Force 0.000003 0.000300 YES
Maximum Displacement 0.000020 0.001800 YES
RMS Displacement 0.000012 0.001200 YES
Predicted change in Energy=-1.312911D-10
Optimization completed.
-- Stationary point found.
----------------------------
! Optimized Parameters !
! (Angstroms and Degrees) !
-------------------------- --------------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------------------
! R1 R(1,2) 1.1923 -DE/DX = 0.0 !
! R2 R(1,3) 1.1923 -DE/DX = 0.0 !
! R3 R(1,4) 1.1923 -DE/DX = 0.0 !
! A1 A(2,1,3) 120.0002 -DE/DX = 0.0 !
! A2 A(2,1,4) 120.0002 -DE/DX = 0.0 !
! A3 A(3,1,4) 119.9997 -DE/DX = 0.0 !
! D1 D(2,1,4,3) 180.0 -DE/DX = 0.0 !
--------------------------------------------------------------------------------
GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
Pseudo-potentials
TlBr3
For TlBrs, the symmetry was constrained to the D3H point group, and the energy and geometry optimised, this time using a LanL2DZ basis set - using pseudo-potentials to model non-valence orbitals on atoms on the second row of the periodic table or below. The Calculation was performed on the HPC.
Result:
- Published to DSpace: DOI:10042/21133
- .log File: JR_TLBR3OPTIMISATION

| Tl-Br Bond Distance / Å | 2.65 |
|---|---|
| Br-Tl-Br Bond angle / ° | 120.0 |
| File Type | .log |
|---|---|
| Calculation Type | FOPT |
| Calculation Method | RB3LYP |
| Basis Set | LANL2DZ |
| Final Energy / au | -91.218 |
| Gradient / au | 0.000 |
| Dipole Moment / D | 0.00 |
| Point Group | D3H |
| Calculation Time / min:sec | 0:38 (On HPC) |
Item Value Threshold Converged?
Maximum Force 0.000002 0.000450 YES
RMS Force 0.000001 0.000300 YES
Maximum Displacement 0.000022 0.001800 YES
RMS Displacement 0.000014 0.001200 YES
Predicted change in Energy=-6.084022D-11
Optimization completed.
-- Stationary point found.
----------------------------
! Optimized Parameters !
! (Angstroms and Degrees) !
-------------------------- --------------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------------------
! R1 R(1,2) 2.651 -DE/DX = 0.0 !
! R2 R(1,3) 2.651 -DE/DX = 0.0 !
! R3 R(1,4) 2.651 -DE/DX = 0.0 !
! A1 A(2,1,3) 120.0 -DE/DX = 0.0 !
! A2 A(2,1,4) 120.0 -DE/DX = 0.0 !
! A3 A(3,1,4) 120.0 -DE/DX = 0.0 !
! D1 D(2,1,4,3) 180.0 -DE/DX = 0.0 !
--------------------------------------------------------------------------------
GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
This bond length was compared to literature values to verify that the calculation had completed without serious errors: literature reports a bond length of 2.55 Å,[1] which, when compared to the calculated result of 2.65 Å, is only a 4% difference, and is therefore plausible.
BBr3
In BBr3, the combination of larger, comre complicated atoms, and smaller, simpler ones led to optimising the molecule by specifying that the Br atoms should be modelled using a pseudo-potential-based LanLDZ basis, and the B using 6-31G(d,p).
Result: JR_BBR3OPTIMISATION

| B-Br Bond Distance / Å | 1.93 |
|---|---|
| Br-B-Br Bond angle / ° | 120.0 |
| File Type | .log |
|---|---|
| Calculation Type | FOPT |
| Calculation Method | RB3LYP |
| Basis Set | Gen |
| Final Energy / au | -64.436 |
| Gradient / au | 0.000 |
| Dipole Moment / D | 0.00 |
| Point Group | D3H |
| Calculation Time / min:sec | 2:05 |
Item Value Threshold Converged?
Maximum Force 0.000008 0.000450 YES
RMS Force 0.000005 0.000300 YES
Maximum Displacement 0.000036 0.001800 YES
RMS Displacement 0.000023 0.001200 YES
Predicted change in Energy=-4.027020D-10
Optimization completed.
-- Stationary point found.
----------------------------
! Optimized Parameters !
! (Angstroms and Degrees) !
-------------------------- --------------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------------------
! R1 R(1,2) 1.934 -DE/DX = 0.0 !
! R2 R(1,3) 1.934 -DE/DX = 0.0 !
! R3 R(1,4) 1.934 -DE/DX = 0.0 !
! A1 A(2,1,3) 120.0 -DE/DX = 0.0 !
! A2 A(2,1,4) 120.0 -DE/DX = 0.0 !
! A3 A(3,1,4) 120.0 -DE/DX = 0.0 !
! D1 D(2,1,4,3) 180.0 -DE/DX = 0.0 !
--------------------------------------------------------------------------------
GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
Results & Bonding analysis
| Molecule | BH3 | BBr3 | TlBr3 |
|---|---|---|---|
| Bond Length / Å | 1.19 | 1.93 | 2.65 |
It is clear that, in this case, the replacement of a Hydrogen substituent with a Bromine ligand results in a longer bond distance, by 0.74 Å, and the replacement of the central Boron with Thallium similarly lengthens the bond - by 0.72 Å. This implies that both the nature of the ligand and of the centre make a difference to the bond nature and strength - and therefore length.
Firstly, the nature of the Hydrogen atom can be compared to that of a Bromine atom - with respect to a Boron centre. Hydrogen is very small, and electron-deficient compared to Bromine. This is likely to result in a much more covalent bond with the Boron centre, as they are similar in electronegativity (Pauling electromnegativities: 2.0 and 2.2 for B and H respectively)[2] and therefore the individual bonds will have very little polarisation. Bromine, on the other hand, is slightly higher in electonegativity (2.9)[2], which will polarise the bond slightly more, lending it ionic character and lengthening it. However, a greater contribution to the longer bond is likely to be the size of the Br orbitals - as Br has 3 filled shells, giving much larger and more diffuse orbitals compared to those of Hydrogen - which has no filled shells, possessing only one electron. In addition, there will be the interaction of the non-bonding Br pz orbitals with the unfilled B pz orbital, leading to some electron donation from the Br, and a bond which, in fact, possesses some π-character, and is therefore longer. This will lead to the empty pz orbitals on the Boron being less available to electron donation, and the BBr3 molecule slightly more stable to lewis bases than the difference in bond length would suggest.
As far as the central atoms are concerned, this data implies a that the Thallium centre will make a shorter bond to the ligand than a Boron, all other things being equal. Boron and Thallium are in the same group, and therefore the same number of electrons are shared with, in this case, Bromine. However, as Thallium much lower down the group, it is bonded very differently. This time, Bromine is much more electronegative than Thallium (1.6)[2], and the bond much more polarised. Leaving the bond closer to being ionic. In addition, the Thallium orbitals, given its position in the periodic table, are much more diffuse and larger than bromine, so the bond is longer and weaker anyway. In addition, the quasi-π interaction with the bromine is gone completely, due to the complete mismatch in size and energy of the pz orbitals on both atoms. Thus the B-Br bond is both shorter and stronger than Tl-Br.
In some cases, Gaussview does not show bonds where, logically,they should be expected. This is because it uses a purely geometrical view of bonds, showing "bond" lines on the model only where the distances are within an expected "bond length" set of distances. This in no way informs that a bond is not present.
Bonds are the effect of overlapping orbitals and of several different forms of electron transfer and sharing. An atom is said to be bonded to another when the distance between them corresponds to an energy minimum from electronic effects. As such, the lengths of them are very variable, and even atoms at what would be considered "extreme" distances share electron density in some manner, and thus can be said to be bonding. However, the limits are not an arbitrary bond/not-bond line - special cases like, for example, Hydrogen-bonding, are not normally shown when drawing a molecule, but they involve electronic attraction between atoms, and make a considerable effect on the geometry and energy of their molecules.
Frequency Analysis
BH3
A frequency, or vibrational analysis was calculated for the 6-31G(d,p)-optimised BH3 molecule.
Result: JR_BH3_OPT_FREQ
| B-H Bond Distance / Å | 1.19 |
|---|---|
| H-B-H Bond angle / ° | 120.0 |
| File Type | .log |
|---|---|
| Calculation Type | FREQ |
| Calculation Method | RB3LYP |
| Basis Set | 6-31G(d,p) |
| Final Energy / au | -26.615 |
| Gradient / au | 0.000 |
| Dipole Moment / D | 0.00 |
| Point Group | C2V |
| Calculation Time / min:sec | 2:39 |
Output
Item Value Threshold Converged?
Maximum Force 0.000005 0.000450 YES
RMS Force 0.000002 0.000300 YES
Maximum Displacement 0.000020 0.001800 YES
RMS Displacement 0.000009 0.001200 YES
Predicted change in Energy=-1.329322D-10
Optimization completed.
-- Stationary point found.
GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
Low frequencies --- -18.6669 -0.0009 -0.0003 0.0006 12.5167 12.5631 Low frequencies --- 1162.9785 1213.1756 1213.2363
Vibration Summary
| # | Vibration Form | Frequency / cm-1 | Intensity | Symmetry (Molecule: D3H) |
|---|---|---|---|---|
| 1 | 
|
1163 | 93 | A2' |
| All B-H bonds stretching symmetrically, all atoms moving up and down z-axis | ||||
| 2 | 
|
1213 | 14 | E' |
| One B-H unit vibrating in x direction, with slight bond vibration, other H atoms rotating symmetrically around z-axis (through x-y plane) | ||||
| 3 | 
|
1213 | 14 | E' |
| One BH2 unit rotating around z-axis (through x-y plane), other H vibrating in y-axis, asymmetrically stretching B-H bond. | ||||
| 4 | 
|
2582 | 0 | A1' |
| All B-H bonds stretching symmetrically out in the x-y plane. B atom is still. | ||||
| 5 | 
|
2715 | 126 | E" |
| 2 B-H bonds stretch out from B atom - opposite motion to each other, other H atom remains still, while B atom oscillates in y axis | ||||
| 6 | 
|
2715 | 126 | E" |
| All B-H bonds stretching - 1 in opposite phase to the other 2, B atom oscillates in x direction |
IR Spectrum

|
| IR Spectrum for BH3 |
|---|
While there are clearly 6 vibrations for BH3, there are only 3 peaks in the IR spectrum. This is because, of those vibrations, there are 2 sets of 2 degenerate vibrations, which contribute to the same peak, not forming separate ones at diffrerent frequencies. In addition, one of the vibrations - the A1' symmetrical stretch, has 0 intensity. It is not IR active because there is no change to the overall dipole moment.
TlBr3
A similar frequency analysis was carried out on TlBr3
Result:
- Published to DSpace: DOI:10042/21176
- .log File: JR_TLBR3_OPT_FREQ
| Tl-Br Bond Distance / Å | 2.65 |
|---|---|
| Br-Tl-Br Bond angle / ° | 120.0 |
| File Type | .log |
|---|---|
| Calculation Type | FREQ |
| Calculation Method | RB3LYP |
| Basis Set | LANL2DZ |
| Final Energy / au | -91.218 |
| Gradient / au | 0.000 |
| Dipole Moment / D | 0.00 |
| Point Group | D3H |
| Calculation Time / min:sec | 0:31 (On HPC) |
Output
Item Value Threshold Converged?
Maximum Force 0.000002 0.000450 YES
RMS Force 0.000001 0.000300 YES
Maximum Displacement 0.000022 0.001800 YES
RMS Displacement 0.000011 0.001200 YES
Predicted change in Energy=-5.660901D-11
Optimization completed.
-- Stationary point found.
GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
Low frequencies --- -3.4213 -0.0026 -0.0004 0.0015 3.9367 3.9367 Low frequencies --- 46.4289 46.4292 52.1449
Vibration Summary
| # | Vibration Form | Frequency / cm-1 | Intensity | Symmetry (Molecule: D3H) |
|---|---|---|---|---|
| 1 | 
|
46 | 4 | E' |
| One Tl-Br unit vibrating in x direction, with slight bond vibration, other Br atoms rotating symmetrically around z-axis (through x-y plane) | ||||
| 3 | 
|
46 | 4 | E' |
| One TlBr2 unit rotating around z-axis (through x-y plane), other Br vibrating in y-axis, asymmetrically stretching Tl-Br bond. | ||||
| 2 | 
|
52 | 6 | A2' |
| All Tl-Br bonds stretching symmetrically, all atoms moving up and down z-axis | ||||
| 4 | 
|
165 | 0 | A1' |
| All Tl-Br bonds stretching symmetrically out in the x-y plane. Tl atom is still. | ||||
| 5 | 
|
211 | 25 | E" |
| 2 Tl-Br bonds stretch out from Tl atom - opposite motion to each other, other Br atom remains still, while Tl atom oscillates in y axis | ||||
| 6 | 
|
211 | 25 | E" |
| All Tl-Br bonds stretching - 1 in opposite phase to the other 2, Tl atom oscillates in x direction |
IR Spectrum

|
| IR Spectrum for TlBr3 |
|---|
Comparison of Vibrational Frequencies
| Vibration Symmetry | BH3 | TlBr3 | ||
|---|---|---|---|---|
| Wavenumber / cm-1 | Intensity | Wavenumber / cm-1 | Intensity | |
| A1' | 2582 | 0 | 165 | 0 |
| A2' | 1163 | 93 | 52 | 6 |
| E' | 1213 | 14 | 46 | 4 |
| E' | 1213 | 14 | 46 | 4 |
| E" | 1275 | 126 | 211 | 25 |
| E" | 1275 | 126 | 211 | 25 |
The frequencies for BH3 and TlBr3 are very different, with those for BH<aub>3 being much higher. This corresponds to much heavier Tl and Br atoms, which therefore vibrate at much lower frequencies.In addition, the vibrational modes themselves are in a different order, in terms of frequency and energy; given that the molecules have the same D3H symmetry, it makes sense that the vibrational modes would be the same.
However, in both spectra, 2 groups of vibrational modes are predicted, the lower-energy A2' and E' modes, and the higher-energy A1' and E" modes. In TlBr3, the order of the higher-energy group is reversed, with the E" modes higher in energy, while, for the lower-energy group, they are all so close together as to not be readily distinguishable on a printed spectrum, and certainly not when calculation error is taken into account. This is most likely because, given the heavier Br atom mass, the deformation into less symmetrical geometries will require more energy than the symmetrical deformations.
It is important to use the same basis set and method for both the optimisation and frequency calculations because the energy values are highly dependent on the method used, and, in fact, cannot be compared at all to those generated by other methods and basis sets, thus, if a frequency analysis is run from an energy corresponding to a different method or basis set, the program will start from a completely different place in its analysis, and therefore the result is extremely likely to be both very different, and highly inaccurate. A frequency analysis both confirms that the optimisation has reached an energy minimum, and gives us the opportunity to predict vibrational modes and therefore the IR spectrum, without handling the actual molecule.
The low frequencies represent the -6 modes in the 3N-6 vibrational modes of a molecule (for which N is the number of atoms). These are the modes corresponding to the centre of mass of the molecule vibrating.
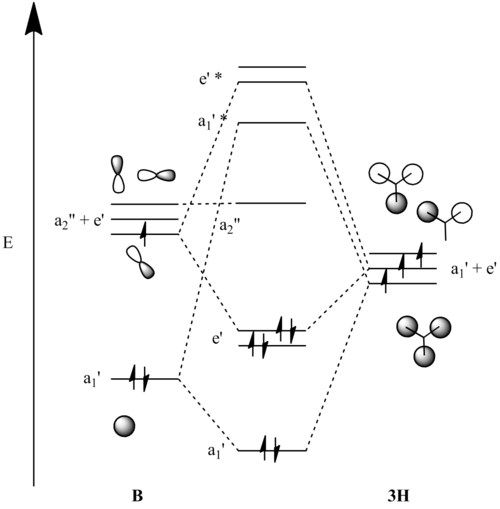
Orbital Analysis
BH3 MOs
A population analysis of the Molecular Orbitals of BH3 was run.
Results: JR_BH3_MOS
| B-H Bond Distance / Å | 1.19 |
|---|---|
| H-B-H Bond angle / ° | 120.0 |
| File Type | .log |
|---|---|
| Calculation Type | SP |
| Calculation Method | RB3LYP |
| Basis Set | 6-31G(d,p) |
| Final Energy / au | -26.615 |
| Gradient / au | 0.000 |
| Dipole Moment / D | 0.00 |
| Point Group | C2V |
| Calculation Time / min:sec | 1:38 |
| MOs for BH3 | |||
|---|---|---|---|
| MO diagram | MO Symmetry | Drawn MO | Calculated MO |
|
e' * |  |

|
| e' * |  |

| |
| a1' * |  |

| |
| a2" |  |

| |
| e' |  |

| |
| e' |  |

| |
| a1' |  |

| |
Clearly, the predicted orbitals bear a reasonable resemblance to the calculated MOS, but the calculations provide a much more accurate picture.
NH3 Optimisation / Frequency
A molecule of NH3 was modelled, optimised and a frequency analysis used to verify that a minimum had been reached, using a B3LYP method and 6-31G(d,p) basis set.
Result: JR_NH3OPTIMISATION

| N-H Bond Distance / Å | 1.02 |
|---|---|
| H-N-H Bond angle / ° | 105.7 |
| File Type | .log |
|---|---|
| Calculation Type | FOPT |
| Calculation Method | RB3LYP |
| Basis Set | 6-31G(d,p) |
| Final Energy / au | -56.558 |
| Gradient / au | 0.000 |
| Dipole Moment / D | 1.85 |
| Point Group | C1 |
| Calculation Time / min:sec | 5:54 |
Output
Item Value Threshold Converged?
Maximum Force 0.000024 0.000450 YES
RMS Force 0.000012 0.000300 YES
Maximum Displacement 0.000079 0.001800 YES
RMS Displacement 0.000053 0.001200 YES
Predicted change in Energy=-1.629727D-09
Optimization completed.
-- Stationary point found.
----------------------------
! Optimized Parameters !
! (Angstroms and Degrees) !
-------------------------- --------------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------------------
! R1 R(1,2) 1.018 -DE/DX = 0.0 !
! R2 R(1,3) 1.018 -DE/DX = 0.0 !
! R3 R(1,4) 1.018 -DE/DX = 0.0 !
! A1 A(2,1,3) 105.7413 -DE/DX = 0.0 !
! A2 A(2,1,4) 105.7486 -DE/DX = 0.0 !
! A3 A(3,1,4) 105.7479 -DE/DX = 0.0 !
! D1 D(2,1,4,3) -111.8631 -DE/DX = 0.0 !
--------------------------------------------------------------------------------
GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
Frequency analysis: JR_NH3FREQUENCY
| N-H Bond Distance / Å | 1.02 |
|---|---|
| H-N-H Bond angle / ° | 105.7 |
| File Type | .log |
|---|---|
| Calculation Type | FREQ |
| Calculation Method | RB3LYP |
| Basis Set | 6-31G(d,p) |
| Final Energy / au | -56.558 |
| Gradient / au | 0.000 |
| Dipole Moment / D | 1.85 |
| Point Group | C1 |
| Calculation Time / min:sec | 3:03 |
Output
Item Value Threshold Converged?
Maximum Force 0.000021 0.000450 YES
RMS Force 0.000009 0.000300 YES
Maximum Displacement 0.000078 0.001800 YES
RMS Displacement 0.000039 0.001200 YES
Predicted change in Energy=-1.611689D-09
Optimization completed.
-- Stationary point found.
GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
<pre>
Low frequencies --- -30.7295 -0.0018 -0.0014 -0.0012 20.1705 28.2664
Low frequencies --- 1089.5535 1694.1244 1694.1856
NH3 NBOs
| NBO Charge Distribution | |
|---|---|

| |
| Range | -1.125 - 1.125 |
| Specific NBO Charge (N) | -1.125 |
| Specific NBO Charge (H) | 0.375 |
Association Energy
A molecule of NH3BH3 was optimised using B3LYP/6-31G(d,p).
Result: JR_NH3BH3OPTIMISATION
| File Type | .log |
|---|---|
| Calculation Type | FOPT |
| Calculation Method | RB3LYP |
| Basis Set | 6-31G(d,p) |
| Final Energy / au | -82.767 |
| Gradient / au | 0.000 |
| Dipole Moment / D | 5.8431 |
| Point Group | C1 |
| Calculation Time / min:sec | 4:48 |
Output
Item Value Threshold Converged?
Maximum Force 0.000086 0.000450 YES
RMS Force 0.000032 0.000300 YES
Maximum Displacement 0.000356 0.001800 YES
RMS Displacement 0.000192 0.001200 YES
Predicted change in Energy=-5.461725D-08
Optimization completed.
-- Stationary point found.
----------------------------
! Optimized Parameters !
! (Angstroms and Degrees) !
-------------------------- --------------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------------------
! R1 R(1,8) 1.0277 -DE/DX = -0.0001 !
! R2 R(2,8) 1.0277 -DE/DX = -0.0001 !
! R3 R(3,8) 1.0277 -DE/DX = -0.0001 !
! R4 R(4,7) 1.212 -DE/DX = -0.0001 !
! R5 R(5,7) 1.212 -DE/DX = -0.0001 !
! R6 R(6,7) 1.212 -DE/DX = -0.0001 !
! R7 R(7,8) 1.6854 -DE/DX = -0.0001 !
! A1 A(4,7,5) 113.5634 -DE/DX = 0.0 !
! A2 A(4,7,6) 113.5634 -DE/DX = 0.0 !
! A3 A(4,7,8) 104.99 -DE/DX = 0.0 !
! A4 A(5,7,6) 113.555 -DE/DX = 0.0 !
! A5 A(5,7,8) 104.9821 -DE/DX = 0.0 !
! A6 A(6,7,8) 104.982 -DE/DX = 0.0 !
! A7 A(1,8,2) 109.3494 -DE/DX = 0.0 !
! A8 A(1,8,3) 109.3494 -DE/DX = 0.0 !
! A9 A(1,8,7) 109.5868 -DE/DX = 0.0 !
! A10 A(2,8,3) 109.347 -DE/DX = 0.0 !
! A11 A(2,8,7) 109.5969 -DE/DX = 0.0 !
! A12 A(3,8,7) 109.5969 -DE/DX = 0.0 !
! D1 D(4,7,8,1) 179.9996 -DE/DX = 0.0 !
! D2 D(4,7,8,2) -60.0015 -DE/DX = 0.0 !
! D3 D(4,7,8,3) 60.0006 -DE/DX = 0.0 !
! D4 D(5,7,8,1) -59.996 -DE/DX = 0.0 !
! D5 D(5,7,8,2) 60.0029 -DE/DX = 0.0 !
! D6 D(5,7,8,3) -179.9949 -DE/DX = 0.0 !
! D7 D(6,7,8,1) 59.9952 -DE/DX = 0.0 !
! D8 D(6,7,8,2) 179.9941 -DE/DX = 0.0 !
! D9 D(6,7,8,3) -60.0038 -DE/DX = 0.0 !
--------------------------------------------------------------------------------
GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
Frequency analysis: JR_NH3BH3FREQUENCY
| File Type | .log |
|---|---|
| Calculation Type | FREQ |
| Calculation Method | RB3LYP |
| Basis Set | 6-31G(d,p) |
| Final Energy / au | -83.225 |
| Gradient / au | 0.000 |
| Dipole Moment / D | 5.56 |
| Point Group | C1 |
| Calculation Time / min:sec | 0:44 |
Output
Item Value Threshold Converged?
Maximum Force 0.000264 0.000450 YES
RMS Force 0.000058 0.000300 YES
Maximum Displacement 0.001470 0.001800 YES
RMS Displacement 0.000376 0.001200 YES
Predicted change in Energy=-2.149033D-07
Optimization completed.
-- Stationary point found.
GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
Low frequencies --- -8.8787 -0.0008 -0.0005 0.0013 19.3581 19.5894 Low frequencies --- 263.3197 631.2464 638.5704
| Molecule | Final Energy / au |
|---|---|
| BH3 | -26.615 |
| NH3 | -56.558 |
| NH3BH3 | -83.225 |
| ΔE | 0.052 |
| ΔE / kJ mol-1 | 137 |
Project: Lewis Acids and Bases
The Calculations required to optimise, carry out a frequency analysis, and view the MOs (of the least energetic) of 4 isomers of Al2Cl4Br2 were run on the HPC, and published to DSpace.
Result:
- Isomer 1
- Optimisation: DOI:10042/21414
- Frequency: DOI:10042/21418
- MOs: DOI:10042/21423
- Isomer 2
- Optimisation: DOI:10042/21415
- Frequency: DOI:10042/21419
- Isomer 3
- Optimisation: DOI:10042/21416
- Frequency: DOI:10042/21421
- Isomer 4
- Optimisation: DOI:10042/21417
- Frequency: DOI:10042/21420