
Rep:Mod:InsertFunnyTitleHere
BH3
The molecule BH3 was optimised with Gaussian. The calculation type was FOPT with method = RB3LYP and basis set 3-21G used. The calculation took 9 sec to run with the final energy reported as -26.46 hartree and a gradient of 0.0002067 a.u which shows that the optimisation found a stationary point/ completed correctly. Initially bond lengths were set to be 1.50 Angstroms long, after optimisation they were found to be 1.19 Angstroms with bond angles of 120.0O. Gaussian correctly identified that the original structure had D3H symmetry limited the final optimisation to only include D3H structures (ie bond angles were fixed) thus the total dipole moment was reported as 0.00 Debye.[1]


Though gaussview only draws in bonds when atoms are close enough together there is a "bond" formed whenever there is a large amount of electron density between atoms thus this is the ideal perameter to corelate display to but in practice this would require a large amount of calualction for each molecule.
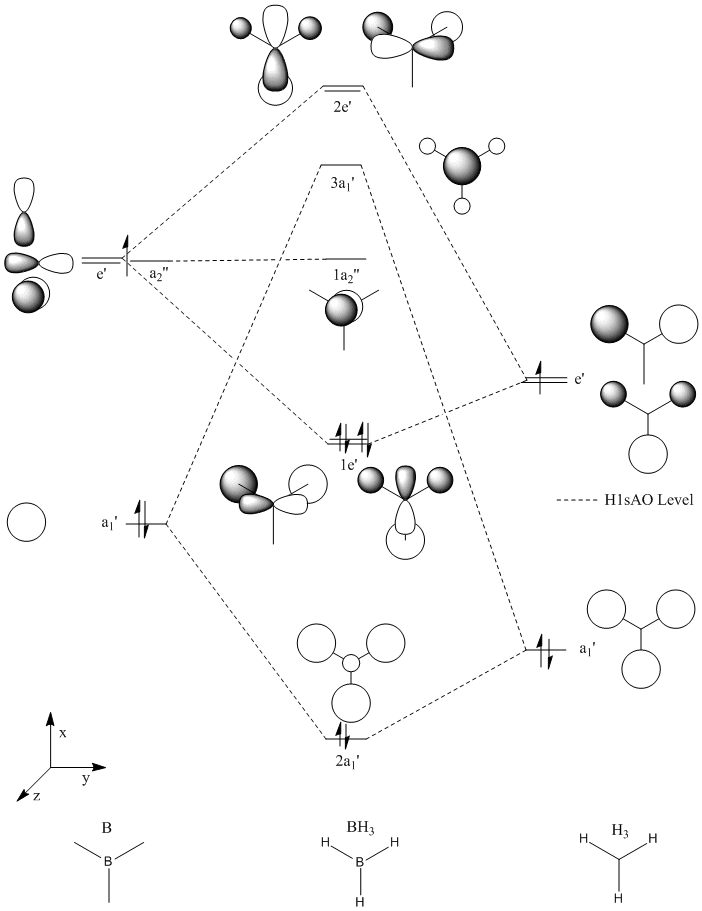
The molecular orbitals of the optimised BH3 molecule shown above were calculated using Gaussian. The calculation was set up with type = SP method = RB3LYP and basis set = 3-21G the extra keywords pop=(full,nbo) were entered. The method and basis set were kept the same as above so that the ground state geometry for the basis set used in the MO calculation was entered. The energy reported for the calculation was the same as the optimisation, this is as expected and shows that there were no errors.
The keyword "pop=(full,nbo)" in the previous calculation invokes a non bonding orbital analysis. From this we see that boron is electron deficient with a non bonding charge of 4.67 making it a lewis base. The hydrogens are all equally electronegative with a charge of 1.11. The analysis also shows us that Boron boron contributes 44.5% of the electron density each bond and is sp2 hybridised. We can also see that one B atomic orbital (that is not core - thus valence) is non bonding and has a negative energy of -0.05430 au, rationalising the lewis acidity of BH3. This orbital is the non-bonding pz orbital which with a symmetry of 1a2 (as seen in the MO diagram above) has no hydrogen atoms to bond with. We can see also that there is very little mixing between MOs.
A frequency analysis was run on the optimised structure calculated above. The calculation was of type = Freq, method = RB2LYP and basis set = 3-21G. The energy reported was exactly the same as for the optimisation showing the calculation completed successfully. Six vibrational frequencies are reported as predicted by the 3N-6 rule. Six low frequencies of significantly lower energy were also reported, that these were so much smaller than the normal frequencies shows that the calculation worked. All of the frequencies reported were positive indicating that the optimisation did not find a transition state. The frequencies output are reported below. A predicted IR spectrum was also generated, as shown below it only contains 5 peaks this is because IR spectroscopy will only detect non-symmetric vibrations as the symmetric vibrations (such the fourth vibration shown below)don't produce a changing dipole moment for radiation to interact with.
 |
|






The optimisation and molecular orbital calculations were run again but this time using method = RB3LYP, Basis set = 6-31G(d) and keywords gfprint & pop(full)[2]. The reported energy for the calculation was -26.613 hartree showing that a better basis set produced a lower energy geometry (bond length still 1.19 Angstroms), though converting between basis sets dosen't work. The rotatable MOs below were the ones produced in this calculation while the static pictures were from the previous calculation.
| 'Bonding' | 'Non-Bonding' | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|







{kind=link}
As can be seen above the orbital shapes predicted by qualitative MO theory match up very well with the rest of the ones calculated as described above (apart from being upside down). This suggests that for small molecules such as the one studied here qualitative diagrams are a good approximation in terms of showing where electron density is located for each orbital but this does not always lead to an accurate prediction of (even relative) energy levels and the diagram did take longer to draw than for the program to run and the method is harder to extrapolate to much larger molecules.
BCl3
A molecule of BCL3 was created and its symmetry was fixed to the D3h point group within a high accuracy of 0.0001. A FOPT optimisation was run on Gaussian with method = RB3LYP and basis set = LANL2MB. LANL2MB indicates the use of basis set of D95V on first row atoms and a pseudo-potential called Los Alamos EPC on heavier atoms. Pseudo-potentials are a method of estimating the effect of larger atoms as they would take too long to compute in the same way as small atoms as they have many more electrons. The calculation terminated with a total energy of -69.43 hartree and a gradient of 0.00005905 au. The Cl-B-Cl bond angles were unsurprisisngly 120.0O with bond lengths of 1.87 Angstroms. The calculation took 8 seconds.
A frequency analysis was carried out on the optimised structure using the same basis set as above to check that a minimum had been found (ie all vibrational frequencies are positive). This was done using the optimised structure and the same basis set as the optimised structure is the most stable (ie minima) only for that particular basis set, if another basis set were used then calculations would essentially be running on an excited state of the molecule. The calculation took 18 seconds. [3]
Gaussview draw in bonds based on interatomic distances, if the gap between atoms is too large then no bond is draw. In unusual (inorganic) molecules bonds are often longer than the cut off number. A bond is essentially an area of high electron density between nuclei of two atoms where the nuclei's attraction to the elections overcomes their repulsion from each other.
The expected symmetry of the BH3 molecule is D3H. For symmetry to be used in the gaussain calculation all of the atoms must be positioned with the exact geometry predicted by the point group. Once the symmetry has been recognised gaussian wont change the molecule out of that point group.
The reported B-Cl bond lengths are 1.87 angstroms this compares to a literature value[4] of 1.82 angstroms. Discrepancy is due to complexation and crystal packing forces present in paper but not the gas phase calculation but shows that calculation is in the right area. The reported Cl-B-Cl bond angles are 120.0 which is fixed by the point group the molecule is fixed to.
Cis&Trans
This section investigates the cis and trans isomers of the octahedral complex MO(CO)4(PCl3)2 shown below. Both isomers were optimised in the following way:
- Initial optimisation using the B3LYP method with the LANM2LB basis set. The additional keyword opt="loose" was added as this is only a rough optimisation.
- The PCl3 groups were next orientated in Gaussview so that the calculation would not get stuck at a local minima. The groups were orientated so that in the trans isomer they eclipsed each other and a CO bond and in the cis isomer so that one cl was aligned with the axial bonds going up and another (in the other group) going down.
- Another optimisation was run again using the B3LYP methiod but a better/larger basis set: LANM2DZ. For this calculation keywords int=ultrafine and scf=conver=9 were used to make the convergence criteria tighter.
- In the final calculation the basis set used on the phosphorus was extended by adding d orbitals. This was achieved adding extrabasis to the keywords list in the .gjf input file and appending the code shown below to the very bottom of this file.
- The results of the calculation are [5] [6] shown below.
(blank line) P 0 D 1 1.0 0.55 0.100D+01 **** (blank line)
| Parameter | Cis | Trans |
|---|---|---|
| Jmol | ||
| Energy\hartree | -623.693 | -623.694 |
There is a small energy gap between isomers of 3.3 kJmol-1 suggesting that the trans isomer is more stable however this gap is smaller than the error (aprox 10kJmol). Calculations carried out in the literature [7](PPh3)2 which suggest that the cis conformer is significantly more stable in the gas phase (as calculated here) and that the main effect causing isomerism to trans is steric clash of the ligands (which should be much smaller in the PCl3 substuited compound. The findings are inline with the empirical evidence [8] that in solution the two isomers readily interconvert, this suggests a low energy difference between the two. The relative stabilities of the cis and trans isomers are result of the trade of between electronics which favour the cis isomer and sterics which favour the trans, however it is reported that [9] solvation effects can easily override this delicate balance. To increase the relative stability of the cis isomer reducing the steric bulk of the lignads would probably have the biggest effect.
A frequency analysis was carried out on the molecules using the extended basis set (one with extra orbitals as described above) on the optimised structure. No negative vibrations were reported for either of the two isomers. Two vibrations were reported for the cis isomer below 45 cm-1 (shown below) no vibrations this low were reported for the trans. Low wavenumber vibrations do not require much energy for the molecule to distort, if they are of an energy comparable to that available at room temperature then they will be constantly vibrating.
| Wavenumber\ cm-1 | Vibration |
|---|---|
| 11 | 
|
| 20 | 
|
Four carbonyl stretching frequencies were reported for both the cis and the trans isomers. All the carbonyl stretches for the cis were expected as all four carbonlys are in slightly different state and were expected to be observed quite strongly as they would all result in a change in dipole moment. Only one CO vibration was expected in the trans isomer as all the carbonlys should have been identical (through rotational symmetry), four peaks were observed as the molecular structure produced by the calcualtions is not exactly symmetrical. This lead to two strongly absorbing vibrations at the same wavenumber and two weekly absorbing ones at higher wavenumbers that were almost symmetric.
References
- ↑ .Log file for BH3 MO analysis [1]
- ↑ .Log file for BH3 MO analysis [2]
- ↑ Log file for calculations of BCl3 [3]
- ↑ Literature Values of B-Cl bond length (in solid sate and as part of an adduct DOI:10.1021/ic50080a032
- ↑ Cis Isomer Calculations [4]
- ↑ Trans Isomer Calculations[5]
- ↑ Dennis W. Bennett, Tasneem A. Siddiquee, Daniel T. Haworth, Shariff E. Kabir and Farzana Camellia. J. Chem. Cryst., 34 (6) (2004) p353-359 DOI:10.1016/S1387-7003(00)00131-3
- ↑ F. Albert Cotton, Donald J. Darensbourg, Simonetta Klein, and Brian W. S. Kolthammer, Inorg. Chem., 21, (1982), p294-299 DOI:10.1023/B:JOCC.0000028667.12964.28
- ↑ Leeni Hirsivaara, Matti Haukka and Jouni Pursiainen, Inorg. Chem. Comm., 3 (2000) p508-510 DOI:10.1021/ic00131a055
Project
My project can be found here