
Rep:Mod:Fr216 Federica
Molecules optimization and analysis
BH3
Computational basis set = 6-31G(d,p)
Ng611 (talk) 15:59, 22 May 2018 (BST) Remember to also add your XC functional to your titles (e.g.: B3LYP)
optimised BH3 molecule |
- The summary table is shown below:

- The item table is shown below:
Item Value Threshold Converged?
Maximum Force 0.000217 0.000450 YES
RMS Force 0.000105 0.000300 YES
Maximum Displacement 0.000919 0.001800 YES
RMS Displacement 0.000441 0.001200 YES
Predicted change in Energy=-1.635270D-07
Optimization completed.
-- Stationary point found.
----------------------------
! Optimized Parameters !
! (Angstroms and Degrees) !
-------------------------- --------------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------------------
! R1 R(1,2) 1.1948 -DE/DX = -0.0002 !
! R2 R(1,3) 1.1944 -DE/DX = -0.0001 !
! R3 R(1,4) 1.1947 -DE/DX = -0.0002 !
! A1 A(2,1,3) 119.9855 -DE/DX = 0.0 !
! A2 A(2,1,4) 120.0157 -DE/DX = 0.0 !
! A3 A(3,1,4) 119.9989 -DE/DX = 0.0 !
! D1 D(2,1,4,3) 180.0 -DE/DX = 0.0 !
--------------------------------------------------------------------------------
- Frequency calculations results:
Low frequencies --- -0.2263 -0.1037 -0.0054 47.9770 49.0378 49.0383 Low frequencies --- 1163.7209 1213.6704 1213.6731
- Frequency analysis log file: FR_BH3_freq.log
Specific additional information
- Vibrations: the vibration table is shown below and are similar to literature values.[1]
| Mode | Frequency | Intensity | Is it IR active? | Symmetry | Type |
|---|---|---|---|---|---|
| 1 | 1164 | 93 | YES | a2 | bend out of plane |
| 2 | 1214 | 14 | YES | e' | bend |
| 3 | 1214 | 14 | YES | e' | bend |
| 4 | 2579 | 0 | NO | a1' | symmetric stretch |
| 5 | 2712 | 126 | YES | e' | asymmetric stretch |
| 6 | 2712 | 126 | YES | e' | asymmetric stretch |
BH3 is a non-linear molecule (D3h point group) and its IR modes can be predicted via the 3N-6 equation.
The IR spectrum does not show 6 peaks because not all the modes are IR active. This is because one of the selection rules for an IR mode to be active is that it requires a change in the dipole moment and mode 4 is totally symmetric causing the dipole moment stays constant. Modes 2,3 and 5,6 are degenerate and appear only once in the spectrum, showing overall only 4 peaks.

- MO analysis

There is no significant difference between the predicted orbitals and the real MOs. That said, the predicted contributions from each fragment to the MO is slightly too big. This implies that the fragments might be closer in energy than the original prediction. The valence electrons are sitting in a bonding orbital, suggesting that the molecule is not willing to give up electrons (act as a base or nucleophile). On the other hand, it has a vacant non-bonding orbital at relatively low energy showing that it is a good electrons acceptor. This not only agrees with the idea that BH3 is electron deficient (VSEPR theory), but also with laboratory experiments where it acts as an acid[3]. MO theory can be useful in predicting orbital interactions and reactivity between fragments in very complex systems, even where the VSEPR or crystal field theories cannot give an explanation.
Ng611 (talk) 16:02, 22 May 2018 (BST) I'm not sure what you mean when you say that the 'predicted contributions from each fragment to the MO is slightly too big'. Do you mean to say that one of the fragments contributes more than predicted, or that both do? More clarity needed here. Besides this, good MO analysis and good discussion.
NH3
Computational basis set = 6-31G(d,p)
optimised NH3 molecule |
- The summary table is shown below:

- The item table is shown below:
Item Value Threshold Converged?
Maximum Force 0.000006 0.000450 YES
RMS Force 0.000004 0.000300 YES
Maximum Displacement 0.000012 0.001800 YES
RMS Displacement 0.000008 0.001200 YES
Predicted change in Energy=-9.844603D-11
Optimization completed.
-- Stationary point found.
----------------------------
! Optimized Parameters !
! (Angstroms and Degrees) !
-------------------------- --------------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------------------
! R1 R(1,2) 1.018 -DE/DX = 0.0 !
! R2 R(1,3) 1.018 -DE/DX = 0.0 !
! R3 R(1,4) 1.018 -DE/DX = 0.0 !
! A1 A(2,1,3) 105.7446 -DE/DX = 0.0 !
! A2 A(2,1,4) 105.7446 -DE/DX = 0.0 !
! A3 A(3,1,4) 105.7446 -DE/DX = 0.0 !
! D1 D(2,1,4,3) -111.8637 -DE/DX = 0.0 !
--------------------------------------------------------------------------------
- Frequency calculation results:
Low frequencies --- -8.5646 -8.5588 -0.0047 0.0454 0.1784 26.4183 Low frequencies --- 1089.7603 1694.1865 1694.1865
- Frequency analysis log file: FR_NH3_freq.log
BH3NH3
Computational basis set = 6-31G(d,p)
optimised BH3NH3 molecule |
- The summary table is shown below:
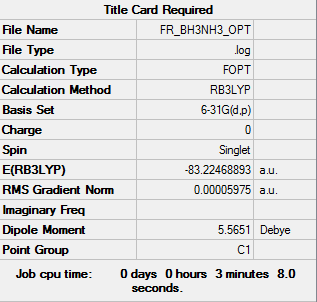

- The item table is shown below:
Item Value Threshold Converged?
Maximum Force 0.000122 0.000450 YES
RMS Force 0.000058 0.000300 YES
Maximum Displacement 0.000513 0.001800 YES
RMS Displacement 0.000296 0.001200 YES
Predicted change in Energy=-1.631175D-07
Optimization completed.
-- Stationary point found.
----------------------------
! Optimized Parameters !
! (Angstroms and Degrees) !
-------------------------- --------------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------------------
! R1 R(1,8) 1.21 -DE/DX = -0.0001 !
! R2 R(2,8) 1.21 -DE/DX = -0.0001 !
! R3 R(3,8) 1.21 -DE/DX = -0.0001 !
! R4 R(4,7) 1.0186 -DE/DX = -0.0001 !
! R5 R(5,7) 1.0186 -DE/DX = -0.0001 !
! R6 R(6,7) 1.0186 -DE/DX = -0.0001 !
! R7 R(7,8) 1.6681 -DE/DX = -0.0001 !
! A1 A(4,7,5) 107.8685 -DE/DX = 0.0 !
! A2 A(4,7,6) 107.8685 -DE/DX = 0.0 !
! A3 A(4,7,8) 111.0294 -DE/DX = 0.0 !
! A4 A(5,7,6) 107.8686 -DE/DX = 0.0 !
! A5 A(5,7,8) 111.0303 -DE/DX = 0.0 !
! A6 A(6,7,8) 111.0302 -DE/DX = 0.0 !
! A7 A(1,8,2) 113.8743 -DE/DX = 0.0 !
! A8 A(1,8,3) 113.8743 -DE/DX = 0.0 !
! A9 A(1,8,7) 104.5972 -DE/DX = 0.0 !
! A10 A(2,8,3) 113.874 -DE/DX = 0.0 !
! A11 A(2,8,7) 104.5968 -DE/DX = 0.0 !
! A12 A(3,8,7) 104.5969 -DE/DX = 0.0 !
! D1 D(4,7,8,1) 179.9998 -DE/DX = 0.0 !
! D2 D(4,7,8,2) -60.0 -DE/DX = 0.0 !
! D3 D(4,7,8,3) 59.9997 -DE/DX = 0.0 !
! D4 D(5,7,8,1) -60.0003 -DE/DX = 0.0 !
! D5 D(5,7,8,2) 59.9998 -DE/DX = 0.0 !
! D6 D(5,7,8,3) 179.9995 -DE/DX = 0.0 !
! D7 D(6,7,8,1) 60.0001 -DE/DX = 0.0 !
! D8 D(6,7,8,2) -179.9998 -DE/DX = 0.0 !
! D9 D(6,7,8,3) -60.0001 -DE/DX = 0.0 !
- Frequency calculation results:
Low frequencies --- -0.0002 0.0004 0.0009 17.6705 28.0331 40.2175 Low frequencies --- 266.5190 632.3638 639.4914
- The frequency log file: BH3NH3_freq.log
Association Energy calculation for BH3NH3
E(NH3)= -56.5778 a.u. = 148545.03 kJ/mol E(BH3)= -26.6153 a.u. = 69878.475 kJ/mol E(NH3BH3)= -83.2247 a.u. = 218506.47 kJ/mol
ΔE=E(NH3BH3)-[E(NH3)+E(BH3)] = -83.2247 -[-56.5778 -26.6153] = -0.0316 a.u. = - 82.96581 kJ/mol
Ng611 (talk) 16:11, 22 May 2018 (BST) Unfortunately you've rounded to too few decimal places for your inputs (should be 5dp instead of 4dp) and you've got an incorrect final value as a result. Take care also to report your answer to only the nearest Kj/mol as these calculations are not accurate beyond that.
The calculated association energy for NH3BH3 is of -82.965 kJ/mol, which corresponds to 82.96581 kJ/mol in terms of dissociation energy. In terms of what kind of bond we are looking at, it is possible to define it as a dative bond by comparison with literature values. The covalent B-N bond has been found to have a dissociation energy of about 380 kJ/mol[4], which is much greater than the valued obtained via the calculations. On the other hand, a dative B-N bond has a dissociation energy of 130.123 ± 4 kJ/mol[5], which is very similar to the calculated value. Therefore, the bond is easily recognizable as a dative bond given by the donation of the N lone pair into the empty B p orbital.[6]
A dative bond is weaker than a covalent bond because while in the first case its rupture gives out species without a net charge (like NH3 and BH3) or two species with both a net charge, the rupture of a covalent bond gives a radical species (far less stable).
This is the reason why BH3NH3 and C2H6 have so different properties despite being isoelectronic[5]. The C-C bond in ethane is covalent and its rupture gives out two radical species (CH3). This is the reason why the C-C bond dissociation energy is so much higher than the B-N one.
BBr3
Computational basis set: B - 6-31G and Br - LanL2DZ
optimised BBr3 molecule |
- The summary table is shown below:

- The item table is shown below:
Item Value Threshold Converged?
Maximum Force 0.000015 0.000450 YES
RMS Force 0.000009 0.000300 YES
Maximum Displacement 0.000058 0.001800 YES
RMS Displacement 0.000042 0.001200 YES
Predicted change in Energy=-1.616527D-09
Optimization completed.
-- Stationary point found.
----------------------------
! Optimized Parameters !
! (Angstroms and Degrees) !
-------------------------- --------------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------------------
! R1 R(1,2) 1.934 -DE/DX = 0.0 !
! R2 R(1,3) 1.934 -DE/DX = 0.0 !
! R3 R(1,4) 1.934 -DE/DX = 0.0 !
! A1 A(2,1,3) 119.9962 -DE/DX = 0.0 !
! A2 A(2,1,4) 120.0034 -DE/DX = 0.0 !
! A3 A(3,1,4) 120.0004 -DE/DX = 0.0 !
! D1 D(2,1,4,3) 180.0 -DE/DX = 0.0 !
- The frequency calculation gave the following results:
Low frequencies --- -4.3208 -2.7694 -2.3007 -0.0002 -0.0001 0.0001 Low frequencies --- 155.8708 155.9431 267.6977
- The frequency log file: BBr3_freq.log
- url for the dspace file: http://hdl.handle.net/10042/202392
DOI:10042/202392
Mini project - Lewis Acids and Bases
The five isomers are shown below, and the label corresponds to their point group.

Confomer with trans-Br groups
Computational basis set: Al, Cl - 6-31G(d,p) and Br - LanL2DZ
optimised trans confomer |
- The summary table for the trans-confomer is shown below:
- The item table is shown below:
Item Value Threshold Converged?
Maximum Force 0.000049 0.000450 YES
RMS Force 0.000019 0.000300 YES
Maximum Displacement 0.000424 0.001800 YES
RMS Displacement 0.000184 0.001200 YES
Predicted change in Energy=-1.664899D-08
Optimization completed.
-- Stationary point found.
----------------------------
! Optimized Parameters !
! (Angstroms and Degrees) !
-------------------------- --------------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------------------
! R1 R(1,3) 2.2982 -DE/DX = 0.0 !
! R2 R(1,4) 2.2982 -DE/DX = 0.0 !
! R3 R(1,5) 2.0936 -DE/DX = 0.0 !
! R4 R(1,6) 2.2746 -DE/DX = 0.0 !
! R5 R(2,3) 2.2982 -DE/DX = 0.0 !
! R6 R(2,4) 2.2982 -DE/DX = 0.0 !
! R7 R(2,7) 2.0936 -DE/DX = 0.0 !
! R8 R(2,8) 2.2746 -DE/DX = 0.0 !
! A1 A(3,1,4) 90.1483 -DE/DX = 0.0 !
! A2 A(3,1,5) 109.8461 -DE/DX = 0.0 !
! A3 A(3,1,6) 110.5133 -DE/DX = 0.0 !
! A4 A(4,1,5) 109.8462 -DE/DX = 0.0 !
! A5 A(4,1,6) 110.5135 -DE/DX = 0.0 !
! A6 A(5,1,6) 121.5158 -DE/DX = 0.0 !
! A7 A(3,2,4) 90.1483 -DE/DX = 0.0 !
! A8 A(3,2,7) 109.8462 -DE/DX = 0.0 !
! A9 A(3,2,8) 110.5135 -DE/DX = 0.0 !
! A10 A(4,2,7) 109.8461 -DE/DX = 0.0 !
! A11 A(4,2,8) 110.5133 -DE/DX = 0.0 !
! A12 A(7,2,8) 121.5158 -DE/DX = 0.0 !
! A13 A(1,3,2) 89.8517 -DE/DX = 0.0 !
! A14 A(1,4,2) 89.8517 -DE/DX = 0.0 !
! D1 D(4,1,3,2) 0.0 -DE/DX = 0.0 !
! D2 D(5,1,3,2) -111.215 -DE/DX = 0.0 !
! D3 D(6,1,3,2) 112.0319 -DE/DX = 0.0 !
! D4 D(3,1,4,2) 0.0 -DE/DX = 0.0 !
! D5 D(5,1,4,2) 111.2149 -DE/DX = 0.0 !
! D6 D(6,1,4,2) -112.0317 -DE/DX = 0.0 !
! D7 D(4,2,3,1) 0.0 -DE/DX = 0.0 !
! D8 D(7,2,3,1) -111.2149 -DE/DX = 0.0 !
! D9 D(8,2,3,1) 112.0317 -DE/DX = 0.0 !
! D10 D(3,2,4,1) 0.0 -DE/DX = 0.0 !
! D11 D(7,2,4,1) 111.215 -DE/DX = 0.0 !
! D12 D(8,2,4,1) -112.0319 -DE/DX = 0.0 !
- Frequency analysis results:
Low frequencies --- -5.3777 -0.0036 -0.0029 -0.0019 1.2979 2.1662 Low frequencies --- 18.1035 49.0942 72.9997
- The frequency log file: Trans confomer.log
MO analysis
The 3 valence MO chosen for the analysis are: MO56, MO54 and MO43.
MO56 is an unfilled orbital. The main orbital interactions are highlighted in blue and the nodes in red. Overall, it has an highly antibonding character.

MO54 is a filled orbital. The main orbital interactions are highlighted in blue and the nodes in red. Overall, it has a non-bonding character.

MO43 is a filled orbital. The main orbital interactions are highlighted in blue. No nodes are present in the MO. Overall, it has an highly bonding character.

Ng611 (talk) 16:17, 22 May 2018 (BST) Your bonding analysis for the last 2 structures is slightly off. MO54 would seem to be to be weakly antibonding and MO43 would be moderately rather than highly antibonding.
Confomer with bridging Br groups
Computational basis set: Al, Cl - 6-31G(d,p) and Br - LanL2DZ
optimised bridging Br confomer |
- The summary table is shown below:

- The item table is shown below:
Item Value Threshold Converged?
Maximum Force 0.000125 0.000450 YES
RMS Force 0.000077 0.000300 YES
Maximum Displacement 0.001576 0.001800 YES
RMS Displacement 0.000754 0.001200 YES
Predicted change in Energy=-6.256738D-07
Optimization completed.
-- Stationary point found.
----------------------------
! Optimized Parameters !
! (Angstroms and Degrees) !
-------------------------- --------------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------------------
! R1 R(1,3) 2.0939 -DE/DX = -0.0001 !
! R2 R(1,5) 2.489 -DE/DX = 0.0001 !
! R3 R(1,6) 2.489 -DE/DX = 0.0001 !
! R4 R(1,8) 2.0929 -DE/DX = 0.0001 !
! R5 R(2,4) 2.0939 -DE/DX = -0.0001 !
! R6 R(2,5) 2.489 -DE/DX = 0.0001 !
! R7 R(2,6) 2.489 -DE/DX = 0.0001 !
! R8 R(2,7) 2.0929 -DE/DX = 0.0001 !
! A1 A(3,1,5) 109.7021 -DE/DX = 0.0001 !
! A2 A(3,1,6) 109.7055 -DE/DX = 0.0 !
! A3 A(3,1,8) 121.7963 -DE/DX = 0.0 !
! A4 A(5,1,6) 91.668 -DE/DX = 0.0001 !
! A5 A(5,1,8) 109.9117 -DE/DX = -0.0001 !
! A6 A(6,1,8) 109.9123 -DE/DX = -0.0001 !
! A7 A(4,2,5) 109.7039 -DE/DX = 0.0001 !
! A8 A(4,2,6) 109.7033 -DE/DX = 0.0001 !
! A9 A(4,2,7) 121.7959 -DE/DX = 0.0 !
! A10 A(5,2,6) 91.6682 -DE/DX = 0.0001 !
! A11 A(5,2,7) 109.9139 -DE/DX = -0.0001 !
! A12 A(6,2,7) 109.911 -DE/DX = -0.0001 !
! A13 A(1,5,2) 88.3319 -DE/DX = -0.0001 !
! A14 A(1,6,2) 88.3319 -DE/DX = -0.0001 !
! D1 D(3,1,5,2) -111.6266 -DE/DX = -0.0001 !
! D2 D(6,1,5,2) 0.0109 -DE/DX = 0.0 !
! D3 D(8,1,5,2) 111.9078 -DE/DX = 0.0 !
! D4 D(3,1,6,2) 111.6235 -DE/DX = 0.0001 !
! D5 D(5,1,6,2) -0.0109 -DE/DX = 0.0 !
! D6 D(8,1,6,2) -111.9073 -DE/DX = 0.0 !
! D7 D(4,2,5,1) -111.6464 -DE/DX = -0.0001 !
! D8 D(6,2,5,1) -0.0109 -DE/DX = 0.0 !
! D9 D(7,2,5,1) 111.8851 -DE/DX = 0.0 !
! D10 D(4,2,6,1) 111.6469 -DE/DX = 0.0001 !
! D11 D(5,2,6,1) 0.0109 -DE/DX = 0.0 !
! D12 D(7,2,6,1) -111.8877 -DE/DX = 0.0 !
- The frequency calculation gave the following results:
Low frequencies --- -5.8862 -5.2899 -4.0205 0.0008 0.0011 0.0029 Low frequencies --- 14.6917 63.2414 86.0249
- The frequency log file: Bridge confomer_freq.log
Difference in energy between confomers
E(isomer with bridging Br groups) = -2352.4063 a.u. E(isomer with trans Br groups) = -2352.4163 a.u.
ΔE = -2352.4063 + 2352.4163 = 0.01 a.u. = 26.255 kJ/mol
Ng611 (talk) 16:22, 22 May 2018 (BST)The final value should be reported to the nearest kj/mol
Both confomers present halogens as bridging units in the dimer. Halogens are generally speaking good bridging groups due to their high electronegativity and presence of lone pair[3]. In the fist case (trans isomer) the bridging atoms are Cl atoms, while in the second they are Br atoms. Cl is more electronegative and less bulky, and gives therefore the more stable ligand (lower energy).
AlCl2Br (monomer)
A computational analysis was performed on the AlCl2Br monomer to get the dissociation energy of the lowest energy dimer.
Computational basis set: Al, Cl - 6-31G(d,p) and Br - LanL2DZ
optimized monomer |
- The summary table is shown below:

- The item table is shown below:
Item Value Threshold Converged?
Maximum Force 0.000136 0.000450 YES
RMS Force 0.000073 0.000300 YES
Maximum Displacement 0.000681 0.001800 YES
RMS Displacement 0.000497 0.001200 YES
Predicted change in Energy=-7.984436D-08
Optimization completed.
-- Stationary point found.
----------------------------
! Optimized Parameters !
! (Angstroms and Degrees) !
-------------------------- --------------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------------------
! R1 R(1,2) 2.089 -DE/DX = -0.0001 !
! R2 R(1,3) 2.089 -DE/DX = -0.0001 !
! R3 R(1,4) 2.2695 -DE/DX = -0.0001 !
! A1 A(2,1,3) 119.8353 -DE/DX = 0.0001 !
! A2 A(2,1,4) 120.0823 -DE/DX = -0.0001 !
! A3 A(3,1,4) 120.0823 -DE/DX = -0.0001 !
! D1 D(2,1,4,3) 180.0 -DE/DX = 0.0 !
- The frequency calculations results are shown below:
Low frequencies --- 0.0023 0.0029 0.0039 1.3569 3.6367 4.2604 Low frequencies --- 120.5042 133.9178 185.895
- The frequency log file: Monomer_freq.log
Dissociation energy calculations
ΔE = - {E(dimer)-[2 x E(monomer)]} = - {- 2352.4163 -[2 x (-1176.1901)]} = 0.0361 a.u. = 94.78056 kJ/mol
The calculation shows how the dimer is lower in energy than the two monomers taken singularly and hence more stable. This is not surprising considering that Al is an electrodeficient Group 13 element, and it tries to compensate by dimerising[3].
References
- ↑ Kawaguchi, Kentarou (1992). "Fourier transform infrared spectroscopy of the BH3 ν3 band". The Journal of Chemical Physics. 96 (5): 3411. doi:10.1063/1.461942. ISSN 0021-9606.
- ↑ P. Hunt, presented in part in "tutorial 1 notes", Imperial College London, May, 2018.
- ↑ 3.0 3.1 3.2 L. Patel, presented in part in "lecture notes", Imperial College London, Term 2, 2018.
- ↑ The table of dissociation energies, accessed Mayb,2018 from: http://staff.ustc.edu.cn/~luo971/2010-91-CRC-BDEs-Tables.pdf
- ↑ 5.0 5.1 Haaland A. Covalent versus Dative Bonds to Main Group Metals, a Useful Distinction. Angew. Chem. Inf. Ed. Engl. 1989;(28)992-1007.
- ↑ The Journal of Chemical Physics 144, 144315 (2016); https://doi.org/10.1063/1.4945624
Ng611 (talk) 16:28, 22 May 2018 (BST) Overall a good report. Remember to report your results to the appropriate amount of accuracy though.