
Rep:Mod:FITF11
The Hydrogenation of Cyclopentadiene Dimer
Two equivalents of 1,3-cyclopentadiene (CPD) dimerises at room temperature to form dicyclopentadiene (DCPD) via a Diels-Alder reaction. One molecule of CPD behaves as the diene, whilst the other as the dienophile. The reaction results in two stereoisomers because the two molecules of CPD can approach each other in two distinct orientations.

The exo- stereoisomer is formed when the substituent (non-reacting double bond) on the dienophile is directed away from the diene whereas the endo- isomer is formed when the substituent is directed towards the diene (Figure 1).
The reaction favours endo-DCPD formation over exo-DCPD (the endo rule). Using 3D modelling programs, Avagrado and ChemBio3D, it was possible using molecular mechanic calculations to determine why this is and whether the reaction is kinetically or thermodynamically controlled.
-
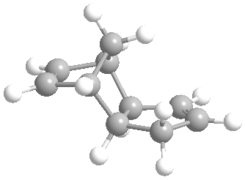 Ball and stick model of Exo-DCPD.
Ball and stick model of Exo-DCPD. -
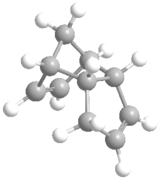 Ball and stick model of Endo-DCPD.
Ball and stick model of Endo-DCPD.


The geometries of both isomers were optimised using Avagrado (Force Field=MMFF94s and Algorithm=Conjugate gradients). The following data was tabulated:
| Property | Exo- (kcal/mol) | Endo- (kcal/mol) |
|---|---|---|
| Stretch | 3.54 | 3.46 |
| Bend | 30.78 | 33.22 |
| Stretch-bend | -2.04 | -2.08 |
| Torsion | -2.73 | -2.95 |
| Van der Waals | 12.79 | 12.33 |
| Electrostatic interactions | 13.01 | 14.18 |
| Dipole moment | 0.31 | 0.74 |
| Total energy | 55.37 | 58.19 |


From Table 1, it is clear that the more thermodynamically stable diastereoisomer is the exo-DCPD because its total energy is lower by 2.82 kcal/mol. This lower energy results from the exo- dimer having lower torsional strain (resistance to bond rotation between atoms that are vicinal) and lower angle bending energy (angle bending between geminal atoms). These relative energy terms represent deviations from "natural" values for the specific function, so the torsional and bending term are deviations from optimal dihedral and bond angles respectively. The bond between the norbornene and cyclopentene rings of the endo-dimer has a greater torsional strain because the two rings are in an eclipsed formation (or are facing each other) to each other. As a result, there is more van der Waals repulsion between the two rings which increases the potential energy of the molecule.
The reaction is kinetically controlled because the kinetic product (endo-) is formed more favourably over the more stable thermodynamic product (exo-). This suggests that the transition state of endo-DCPC must be lower (lower activation energy, greater rate constant) therefore it forms more quickly than exo-DCPD.
By studying the frontier orbitals (HUMO and LUMO orbitals) of endo-DCPD (Figure 3) it is apparent the back orbitals of the diene have the correct symmetry for favourable interactions with the back orbitals of the dieneophile. Although no bonds form between these orbitals, this favourable through space interaction stabilises the transition state and therefore lowers its energy hence endo-DCPD is the kinetic product.
A single step hydrogenation of DCPD breaks a single double bond but because there are two distinct double bonds, the reaction is regioselective. The hydrogenation can either take place in the cyclopentene ring to yield dimer A or in the norbornene ring to yield dimer A. (Figure 4.)

Again, using molecular mechanical calculations, the relative ease of hydrogenation at each specific double bond can be obtained. The following data was tabulated:
| Property | Dimer A (kcal/mol) | Dimer B (kcal/mol) |
|---|---|---|
| Stretch | 3.31 | 2.82 |
| Bend | 30.85 | 24.68 |
| Stretch-bend | -1.93 | -1.66 |
| Torsion | 0.07 | -0.38 |
| Van der Waals | 13.28 | 10.64 |
| Electrostatic interactions | 5.12 | 5.15 |
| Dipole moment | 0.51 | 0.41 |
| Total energy | 50.72 | 41.26 |
Studying Table 2, dimer B is lower in total energy compared to dimer A by 9.46 kcal/mol thus dimer B is more thermodynamically stable. This difference in total energy arises from the bending term (as discussed previously this is the deviation from optimal bond angles).
-
 Figure 5: Dimer A showing bond angle of alkene in the norbornene ring.
Figure 5: Dimer A showing bond angle of alkene in the norbornene ring. -
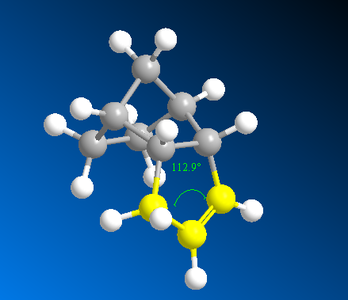 Figure 6: Dimer B showing bond angle of alkene in the cyclopentene ring.
Figure 6: Dimer B showing bond angle of alkene in the cyclopentene ring.


The optimal bond angle for an alkene is 120° but as shown clearly by Figure 5 and 6, both the bond angles for the alkene in dimer A and B are not at optimal, thus producing strain on the double bond. However, the bond angle for the double bond in dimer B is closer to optimal than in dimer A therefore is less strained. As a result, the double bond (being more strained) in the norbornene ring is more likely to break during hydrogenation to form dimer B.