
Rep:Mod:1234Abcd
Synthesis and Computational lab: 1C
Submitted by William Lim
Conformational analysis using Molecular Mechanics
The Hydrogenation of Cyclopentadiene Dimer
Avogadro was used to define the two products 1 and 2 and their geometries were optimized using the MMFF94(s) force field option. The two products of hydrogenation 3 and 4 were similarly compared so that a thermodynamic prediction of the relative ease of hydrogenation of each of the double bonds in 2 could be obtained.
optimized structure of 1
optimized structure of 2
optimized structure of 3
optimized structure of 4
The energies produced by the MMFF94(s) program for the molecules are shown below:
| 1 | 2 | 3 | 4 | |
|---|---|---|---|---|
| Total bond stretching energy (kcal/mol) | 3.54317 | 3.46778 | 3.31219 | 2.82329 |
| Total angle bending energy (kcal/mol) | 30.77257 | 33.18837 | 31.93349 | 24.68546 |
| Total torsional energy (kcal/mol) | -2.73097 | -2.94986 | -1.46888 | -0.37878 |
| Total van der Waals energy (kcal/mol) | 12.80164 | 12.35964 | 13.63879 | 10.63743 |
| Total electrostatic energy (kcal/mol) | 13.01367 | 14.18510 | 5.11946 | 5.14701 |
| Total energy (kcal/mol) | 55.37342 | 58.19067 | 50.44567 | 41.25749 |
The energy of 2 is higher than the energy of 1. This suggests that the Diels-Alder reaction is under kinetic control because the exo product is more stable yet the endo product is produced specifically.1
Since the energy of 3 is significantly higher than the energy of 4, it is easier to hydrogenate the double bond in 4 from a thermodynamic point of view.
There is a more positive angle bending energy for 3 compared to 4 which indicates that the predicted bonds in 3 are further from the "natural" angles compared to 4.
Avogadro was used to define the two products 9 and 10 and their geometries were optimized using the MMFF94(s) force field option.The results are shown below:
optimized structure of 9
optimized structure of 10
| 9 | 10 | |
|---|---|---|
| Total bond stretching energy (kcal/mol) | 13.62169 | 7.60020 |
| Total angle bending energy (kcal/mol) | 45.58605 | 18.78765 |
| Total torsional energy (kcal/mol) | 18.82357 | 0.17972 |
| Total van der Waals energy (kcal/mol) | 46.71011 | 33.35319 |
| Total electrostatic energy (kcal/mol) | 1.85688 | -0.05751 |
| Total energy (kcal/mol) | 128.94158 | 60.56433 |
The results suggest that the conformation of 10 is more stable than the conformation of 9. 10 seems to be far more stable than 9. Perhaps I have not optimized the geometry of 9 to its minimum.
The conformation of the cyclohexane ring in 9 and 10 was changed to minimize the energy further. Optimizing geometries of the whole molecule using the MMFF94(s) force field option does not optimize to the minimum energy. Atoms sometimes have to be adjusted to a conformation that is similar to the conformation of the molecule at minimum energy. My attempts at optimizing the positions of the atoms on the cyclohexane ring seem to suggest that the chair conformation of the cyclohexane ring gives the lowest energy conformation for the whole molecule.
The alkene in 9 and 10 would be expected to react slowly according to Maier and Schleyer. They suggest that ‘hyperstable olefins are stabilized rather than destabilized because of their location at a bridgehead and should be thermodynamically more stable than any of their positional isomers. Hyperstable olefins should be remarkably unreactive'. However, this was only the result of analyzing calculations using Allinger’s MM1 empirical force field program.2
Spectroscopic Simulation using Quantum Mechanics
The 1H and 13C NMR spectra of molecules 17 and 18 were simulated and compared with literature values3 to see if the literature values have been correctly interpreted and assigned.
To simulate their spectra, the molecules were optimized. Avogadro was used to define the two products 17 and 18 and their geometries were optimized using the MMFF94(s) force field option. The results are shown below:
optimized structure of 17
optimized structure of 18
| 17 | 18 | |
|---|---|---|
| Total bond stretching energy (kcal/mol) | 15.86780 | 15.05778 |
| Total angle bending energy (kcal/mol) | 31.30292 | 30.73119 |
| Total torsional energy (kcal/mol) | 13.56290 | 9.74799 |
| Total van der Waals energy (kcal/mol) | 53.68769 | 49.53099 |
| Total electrostatic energy (kcal/mol) | -6.97045 | -6.08148 |
| Total energy (kcal/mol) | 109.06600 | 100.56495 |
According to these energies, the conformation of 18 is less strained than the conformation of 17.
The conformation shown in the literature3 for molecule 17 is not clear. The cyclohexane ring in that structure seems to adopt a twist boat structure and in my calculations has a higher energy (109.06600 kcal/mol). I reoptimized 17 so that the cyclohexane ring had a chair conformation. As expected, the energy of this reoptimized 17 molecule was lower:
| reoptimized 17 | |
|---|---|
| Total bond stretching energy (kcal/mol) | 15.90062 |
| Total angle bending energy (kcal/mol) | 31.44611 |
| Total torsional energy (kcal/mol) | 11.26328 |
| Total van der Waals energy (kcal/mol) | 51.91029 |
| Total electrostatic energy (kcal/mol) | -7.39755 |
| Total energy (kcal/mol) | 104.63008 |
The calculated and literature 17 C NMR Shifts (for 17 with total energy: 109.06600 kcal/mol) are shown below:
| Carbon Atom | Predicted/Computed Chemical Shift (ppm) | Experimental Chemical Shift (ppm) | Absolute Difference (ppm) |
|---|---|---|---|
| 7 | 217.12 | 218.79 | 1.67 |
| 4 | 145.12 | 144.63 | 0.49 |
| 9 | 125.47 | 125.33 | 0.14 |
| 14 | 89.10 | 72.88 | 16.22 |
| 12 | 59.09 | 56.19 | 2.9 |
| 13 | 54.40 | 52.52 | 1.88 |
| 3 | 53.43 | 48.50 | 4.93 |
| 5 | 51.88 | 46.80 | 5.08 |
| 6 | 47.74 | 45.76 | 1.98 |
| 15 | 47.63 | 39.80 | 7.83 |
| 21 | 44.18 | 38.81 | 5.37 |
| 22 | 42.50 | 35.85 | 6.65 |
| 11 | 38.64 | 32.66 | 5.98 |
| 18 | 35.45 | 28.79 | 6.66 |
| 1 | 32.23 | 28.29 | 3.94 |
| 17 | 28.44 | 26.88 | 1.56 |
| 2 | 26.39 | 25.66 | 0.73 |
| 24 | 26.26 | 23.86 | 2.4 |
| 16 | 21.46 | 20.96 | 0.5 |
| 25 | 19.74 | 18.71 | 1.03 |
The calculated reoptimized 17 and literature 17 C NMR Shifts are shown below:
| Carbon Atom | Predicted/Computed Chemical Shift (ppm) | Experimental Chemical Shift (ppm) | Absolute Difference (ppm) |
|---|---|---|---|
| 7 | 216.27 | 218.79 | 2.52 |
| 4 | 144.86 | 144.63 | 0.23 |
| 9 | 124.71 | 125.33 | 0.62 |
| 14 | 91.35 | 72.88 | 18.47 |
| 12 | 60.96 | 56.19 | 4.77 |
| 13 | 57.08 | 52.52 | 4.56 |
| 3 | 52.46 | 48.50 | 3.96 |
| 5 | 51.48 | 46.80 | 4.68 |
| 6 | 46.92 | 45.76 | 1.16 |
| 21 | 43.87 | 39.80 | 4.07 |
| 22 | 41.94 | 38.81 | 3.13 |
| 15 | 41.87 | 35.85 | 6.02 |
| 11 | 35.43 | 32.66 | 2.77 |
| 1 | 31.19 | 28.79 | 2.4 |
| 17 | 28.94 | 28.29 | 0.65 |
| 18 | 28.36 | 26.88 | 1.48 |
| 2 | 27.37 | 25.66 | 1.71 |
| 24 | 26.33 | 23.86 | 2.47 |
| 16 | 24.55 | 20.96 | 3.59 |
| 25 | 19.77 | 18.71 | 1.06 |
This reoptimized conformation of 17 seems to be a better fit to the literature values than 17 (with total energy: 109.06600 kcal/mol).3 This suggests that the conformation of the cyclohexane ring shown in the literature for 17 is misleading.
The calculated and literature 18 C NMR Shifts are shown below:
| Carbon Atom | Predicted/Computed Chemical Shift (ppm) | Experimental Chemical Shift (ppm) | Absolute Difference (ppm) |
|---|---|---|---|
| 7 | 212.23 | 211.49 | 0.74 |
| 4 | 148.08 | 148.72 | 0.64 |
| 9 | 120.14 | 120.90 | 0.76 |
| 14 | 94.11 | 74.61 | 19.5 |
| 12 | 60.42 | 60.53 | 0.11 |
| 3 | 54.88 | 51.30 | 3.58 |
| 13 | 54.20 | 50.94 | 3.26 |
| 5 | 49.69 | 45.53 | 4.16 |
| 15 | 49.14 | 43.28 | 5.86 |
| 22 | 46.77 | 40.82 | 5.95 |
| 21,17 | 41.79 | 38.73 | 3.06 |
| 6 | 38.74 | 36.78 | 1.96 |
| 11 | 34.13 | 35.47 | 1.34 |
| 18 | 33.64 | 30.84 | 2.8 |
| 2 | 28.15 | 30.00 | 1.85 |
| 24 | 26.46 | 25.56 | 0.9 |
| 1 | 24.54 | 25.35 | 0.81 |
| 25 | 22.62 | 22.21 | 0.41 |
| 16 | 21.71 | 21.39 | 0.32 |
Carbons near the Sulphur atoms have significantly different calculated and experimental chemical shifts. This is due to spin-orbit coupling errors. The carbonyl group seems to have a small effect on the chemical shifts too, but far less than the effect of the sulphur atoms.
The H NMR spectra are more difficult to analyze. It would be easier if the spin-couplings of 17 and 18 were calculated using the keyword: # b3lyp/6-311+G(d,p) scrf(cpcm,solvent=chloroform) NMR(spinspin,mixed) and the simulated observed NMR was calculated.
Attempts at comparing the H NMR spectra are shown below:
17 (not reoptimized) H NMR Shifts are shown below:
| Carbon Atom | Predicted/Computed Chemical Shift (ppm) | Experimental Chemical Shift (ppm) |
|---|---|---|
| 10 | 5.15 | Not in experiment |
| Not in calculation | 4.84 | |
| 45 | 3.45 | Not in experiment |
| 46, 44, 47 | 3.30, 3.21 | 3.40-3.10 |
| 34, 19 | 3.02 | Not in experiment |
| Not in calculation | 2.99 | |
| 33, 51, 36, 31, 35, 28, 32, 26, 29, 39, 30, 38, 27, 37, 42, 48, 40 | 2.66, 2.55, 2.49, 2.29, 2.15, 2.08, 1.72, 1.66, 1.58, 1.53, 1.35 | 2.80-1.35 |
| Not in calculation | 1.38 | |
| Not in calculation | 1.25 | |
| 43 | 1.11 | 1.10 |
| 52,49,50,41,53 | 0.88 | 1.00-0.80 |
18 H NMR Shifts are shown below:
| Carbon Atom | Predicted/Computed Chemical Shift (ppm) | Experimental Chemical Shift (ppm) |
|---|---|---|
| 10 | 5.98 | Not in experiment |
| Not in calculation | 5.21 | |
| 45,46 | 3.19 | Not in experiment |
| 47 | 3.06 | Not in experiment |
| 44, 31,33,28 | 2.96, 2.77 | 3.00-2.70 |
| 34, 27,36,19 | 2.65, 2.48 | 2.70-2.35 |
| 43 | 2.33 | Not in experiment |
| 37 | 2.23 | Not in experiment |
| 29,35,30, 26 | 2.00, 1.84 | 2.20-1.70 |
| 51,32,48,39 | 1.58 | 1.58 |
| 41,40,38, 52 | 1.27, 1.20 | 1.50-1.20 |
| Not in calculation | 1.07 | |
| Not in calculation | 1.03 | |
| 53,50,49 | 0.96 | Not in experiment |
| 42 | 0.63 | Not in experiment |
It is clear that the calculated results (not corrected for spin-orbit coupling errors) do not agree with the literature values. Furthermore, calculating the observed H NMR spectra would take a lot of time. Obtaining the calculated C NMR values to compare to C NMR literature values is a faster process.
For 17: the sum of electronic and thermal Free Energies is -1651.451315 Hartrees. (File:17.log) (17 C NMR) (17 H NMR)
{kind=link}
{kind=link}
For 17 reoptimized: the sum of electronic and thermal Free Energies is -1651.460771 Hartrees. (File:17 reoptimized on 5 Mar.log) (17 reoptimized C NMR) (17 reoptimized H NMR)
{kind=link}
{kind=link}
For 18: the sum of electronic and thermal Free Energies is -1651.463260 Hartrees. (File:18.log) (18 C NMR) (18 H NMR)
{kind=link}
{kind=link}
18 has a lower energy than 17 and this suggests that it is more stable than 17.
Analysis of the properties of the synthesised alkene epoxides
The crystal structures of the two catalysts
The Shi and Jacobsen catalyst crystal structures were found using the Conquest program.
Shi Catalyst
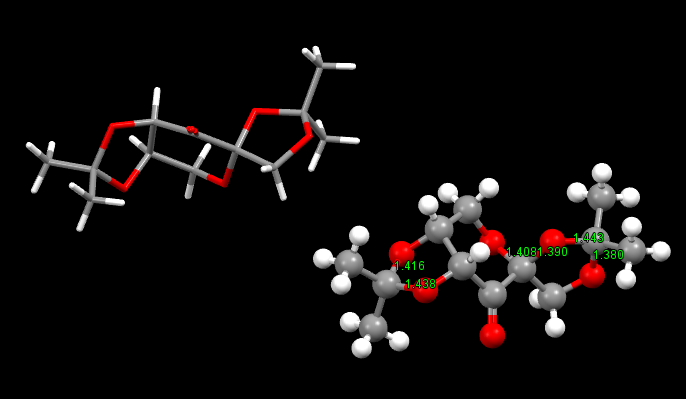
The crystal structure and C-O bond lengths for anomeric centres are shown below:
(note that two molecules of the catalyst are shown in the .mol file and image)
Crystal structure of Shi catalyst
C-O bond lengths for anomeric centres
{kind=link}
The tetrahydropyran ring in the catalyst seems to exhibit the anomeric effect. The heteroatom (oxygen atom) as the substituent is in an axial position.
Jacobsen Catalyst
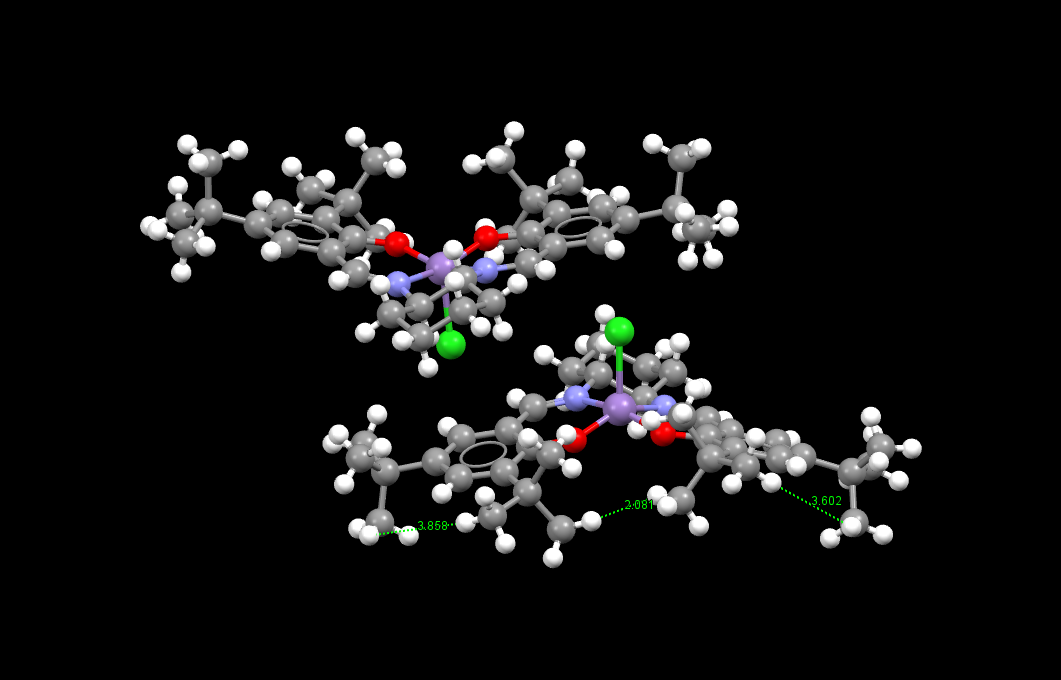
The t-Bu groups in the catalyst seem to be repelling each other; there seem to be through-space steric interactions between all of the t-Bu groups which minimize the steric hindrance in the conformation of the catalyst. This is shown in the Mercury program but not in the .mol file. The distances between hydrogens on adjacent t-Bu groups were investigated to see how close they were:
Crystal structure of Jacobsen catalyst
Distances between hydrogens on adjacent t-Bu groups
{kind=link}
(note that two molecules of the catalyst are shown in the .mol file and image)
The atoms directly bonded to the transition metal and the transition metal adopt a distorted square pyramidal geometry. This is perhaps due to the conformation of the salen ligand. The chlorine atom position is something not expected.
The calculated NMR properties of your products
The C and H NMR spectra for 10 conformations were calculated. The NMR data are shown below:
Assigning the absolute configuration of the product
The reported literature for optical rotations
| Conformation | Optical Rotatory power (°) | Comments | Reference |
|---|---|---|---|
| 1,2-(R,S)-Dihydronaphthalene oxide | -144.9 | Conc: 0.33 g/100mL; Solv: chloroform (67-66-3); Wavlen: 589.3 nm; Temp: 25 °C | 4 |
| (R)-Styrene oxide | -24.0 | Conc: 1.00 g/100mL; Solv: chloroform (67-66-3); Wavlen: 589.3 nm; Temp: 21 °C |
5 |
| (S)-Styrene oxide | +22.8 | Conc: 0.98 g/100mL; Solv: chloroform (67-66-3); Wavlen: 589.3 nm; Temp: 24 °C |
6 |
| (R,R)-trans-Stilbene oxide | +250.8 | Conc: 0.85 g/100mL; Solv: chloroform (67-66-3); Wavlen: 589.3 nm; Temp: 22 °C |
7 |
| (-)-(S,S)-trans-Stilbene oxide | -251 | Conc: 5.82 g/100mL; Solv: ethanol (64-17-5); Wavlen: 589.3 nm | 8 |
| (R,R) trans-β-methylstyrene oxide | +40.8 | Conc: 0.92 g/100mL; Solv: chloroform (67-66-3); Wavlen: 589.3 nm; Temp: 25 °C | 9 |
| (S,S) trans-β-methylstyrene oxide | -41.7 | Conc: 0.88 g/100mL; Solv: chloroform (67-66-3); Wavlen: 589.3 nm; Temp: 24 °C | 10 |
| (S,R) cis-β-methylstyrene oxide | +17.7 | Conc: 0.94 g/100mL; Solv: chloroform (67-66-3); Wavlen: 589.3 nm; Temp: 25 °C |
9 |
The calculated chiroptical properties of the product
| Conformation | Optical Rotatory power (°) | DOI for calculation |
|---|---|---|
| 1,2-(R,S)-Dihydronaphthalene oxide | -35.86 | DOI:10042/27970 |
| 1,2-(S,R)-Dihydronaphthalene oxide | +35.86 | DOI:10042/27969 |
| (R)-Styrene oxide | -30.30 | |
| (S)-Styrene oxide | +30.14 | DOI:10042/27968 |
| (R,R)-trans-Stilbene oxide | +298.33 | DOI:10042/27972 |
| (-)-(S,S)-trans-Stilbene oxide | -297.77 | DOI:10042/27971 |
The literature value of the optical rotation of 1,2-(R,S)-Dihydronaphthalene oxide differs significantly from the calculated optical rotation. Perhaps the conformation of the structure used to calculate the optical rotation is wrong.
There are often many literature values of optical rotatory power for a molecule. The angle depends on the type of solvent used, concentration of sample, the wavelength of incident light, and temperature. However, only one reported value of an optical rotatory power is calculated for each conformation of epoxide using the method in experiment 1C, and only the solvent and wavelength are known. It is thus difficult to compare the calculated values to the literature values because the concentration and temperature are not known.
Using the (calculated) properties of transition state for the reaction (β-methyl styrene only)
The diastereomeric transition state with the lowest free energy (Sum of electronic and thermal Free Energies) was used for each conformation compared. The free energy difference between two diastereomeric transition states was then calculated. This was originally in units of Hartrees and was subsequently converted into Joules/mol. These differences were then converted into K (using the equation: Gibbs free energy = -RTlnK, where T = 298.15 K) and ee.
The results are shown below:
Jacobsen catalyst:
reaction 1: trans-β-methyl styrene: ratio of R,R to S,S or S,S to R,R: K= 5533.056; ee= 100%
reaction 2: cis-β-methyl styrene: ratio of S,R to R,S or R,S to S,R: K = 8118.628; ee= 100%
Shi catalyst:
reaction 3: trans-β-methyl styrene: ratio of R,R to S,S or S,S to R,R: K = 3486.647; ee= 100%
This would suggest that 100% of one enantiomer is exclusively produced for each of these three reactions.
The equation ee (%) = (R-S)/(R+S) * 100 was used.
Note: I am not sure if the calculated K and ee values are correct.
Investigating the non-covalent interactions in the active-site of the reaction transition state
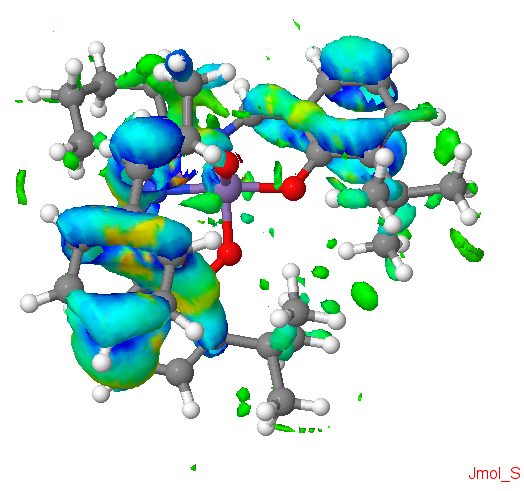
The transition state associated with the reaction of the Jacobsen catalyst and S,S trans-β-methyl styrene was investigated.
NCIs in the transition state (reaction of the Jacobsen catalyst and S,S trans-β-methyl styrene)
{kind=link}
Interesting interactions are the the NCIs between the aromatic ring of the reactant and the aromatic ring of the salen ligand. There are also NCIs between the nitrogen atom of the salen ligand and the reactant. These strong attractions, indicated by the blue colour, could explain the origin of the stereoselectivity of the reaction.
References
- J. Claydon, N. Greeves, S. Warren and P. Wothers, in Organic Chemistry, Oxford University Press, New York, 2001, p. 912.
- W. F. Maier and P. R. Schleyer, J. Am. Chem. Soc., 1981, 103, 1891-1900.
- L. A. Paquette, N. A. Pegg, D. Toops, G. D. Maynard, and R. D. Rogers, J. Am. Chem. Soc., 1990, 112, 277-283.
- Page, Philip C. Bulman; J. Org. Chem., 2007, 72, 4424-4430.
- D. C. Forbes, Tetrahedron, 2008, 65, 70-76.
- K. Matsumoto, Angewandte Chemie, International Edition, 2009, 48, 7432-7435.
- D. J. Fox, Organic & Biomolecular Chemistry, 2006, 4, 3117-3119.
- T. Saito, ARKIVOC (Gainesville, FL, United States), 2004, 2, 152-171.
- B. Wang, J. Org. Chem., 2009, 74, 3986-3989.
- H. Tanaka, J. Am. Chem. Soc., 2010, 132, 12034-12041.