Retrieving an NMR data file from the Jeol Spectrometer
On Windows 10, type run into the search box at the bottom
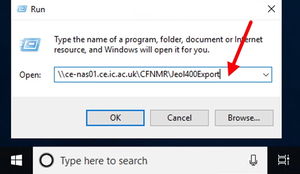
step 2In the run box, type \\ce-nas01.ce.ic.ac.uk\CFNMR\Jeol400Export\chemteaching2\nmr\ and find your own file as per issued instructions.
step 3Now, from the Software Hub panel, firstly select a chemistry filter
If you cannot see anything relating to the software hub, open a Web Browser and type https://softwarehub.imperial.ac.uk/login as the URL and proceed to log in using your Imperial Credentials
step 4From the list of chemistry programs (32) invoke MestreNova. Use the latest version (14). Drag-n-drop the Jeol data .jdf data file into the MestreNova empty window
step 5A spectrum will appear.
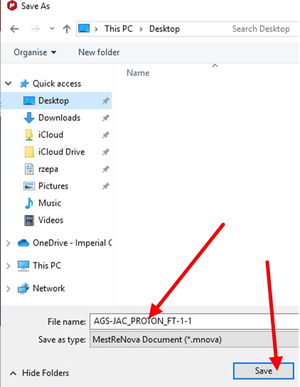
step65When you have finished inspecting the spectrum, save the data file using Save as as a .mnova file and repeat for .jdx format. Take care to note where you are saving the files (on the desktop in this example).
OPTIONAL: As an alternative to 3-6 above, a brand new NMR analysis tool has recently appeared: https://www.nmrium.org/ In many ways this is simpler than using MestreNova but achieves the same result. Give it a go and let us know if you prefer it to Mnova!
You will now have FIVE files you need to make your data deposition. These are:
a .jdf original data file,
an .mnova original data + processed spectrum document
a .jdx file in JCAMP_DX formt
a .cdxml chemdraw document you prepared earlier
a .pdf Acrobat file.
Crib sheet for Processing a spectrum using MestreNova
These are some short hints for how to quickly get the most out of your data.
Once you have opened the NMR folder using MN, make sure the tab along the top of the program is showing NMR/Analysis
Go to Chemdraw, select the structure of your molecule and copy it/paste it into the Mnova Window. This will serve to remind you of the molecule
From 1H Spectrum in the second row of icons, select compound peaks. This will attempt to remove well known solvent peaks such as CHCl3 or H2O from your spectrum, which may help with subsequent integrations. You can also show only the solvents/Impurities peaks for information.
Using the toolbar on the right of the program, find the Increase intensity icon to make the spectrum higher
Using the toolbar on the right of the program, select the zoom in icon at the top and drag from the leftmost peak to the rightmost peak to expand the spectrum.
In the top NMR/Analysis icon collection, click on Manual integrate and select th left and right edges of a peak that you think you can identify. For example, an aromatic group or a methyl group.
An integral curve will appear and below the peak a numerical integration of 1.00 will appear.
Place the cursor on the horizontal line just above the numeric integration. Both the number and the integration curve go red. Now right click whilst the red is still showing.
Select Edit integral and normalize to what you think the integral should correspond to. Thus methyl = 3, a monosubstituted phenyl = 5, a methylene group = 2 etc.
Optionally, select linear correction and then autocorrect. This might improve the baseline and hence the integration.
You can now inspect the closest integer for each peak. This will only be approximate. It will also give the total integration which should correspond to the number of protons in your molecule. Obviously if your compound is a mixture of several molecules, you might not get integers for the peaks!
At this stage you should save what you have obtained as a .mnova file
Only if you are keen, select the next tab in NMR/Assignments. You can now select a peak and assign it to a proton on the chemdraw diagram. Save the file again.