
Organic:whogotthefunk
The Hydrogenation of the Cyclopentadiene Dimer


The cyclopentadiene dimer can be in one of two forms - either molecule 1 (see diagram on the right) or molecule 2. They are endo-exo isomers of one another - molecule 1 being exo and molecule 2 being endo. To work out which of the two is more stable, both molecules weremodelled in ChemBio3D Ultra, and was then geometrically optimised. Molecule 1 was done first, and this gave a value of 133.6 kJ/mol. Although this number has little value when taken on its own, it can be used to make comparisons between the two isomers of this molecule.
Due to the steric hindrance present in molecule 2 (caused by the two hydrogen atoms clashing with the bridging carbons), it seems likely that molecule 1 will have a lower energy. This is confirmed by the result, which gives molecule 2 a value of 142.4 kJ/mol, showing that molecule 1 is the more energetically favoured of the two - and therefore the thermodynamic product (meaning that molecule 2, shown to the left, is the kinetic product). Given that it is only molecule 2 that is produced in the reaction, the reaction is therefore kinetically controlled.
ChemBio3D can also be used to predict the ease at which each of the double bonds in molecule 2 can be hydrogenated. Molecules 3 and 4, shown in the diagram to the right, show the two possible variations of molecule 2 when it contains one double bond (and has therefore been hydrogenated once). To determine which of the two products is more stable, and thus more likely to be the primary hydrogenation product, each of the two molecules were drawn in ChemBio3D Ultra and then geometrically optimised. Other data, such as the hydrogen bond strength, was also obtained, and this is shown in the table below (all units in the table in kJ/mol).

By comparing the total energies of the two molecules, it can be seen that molecule 4 has the lower total energy of the two, and is therefore more stable than molecule 3 - showing that molecule 4 would be favoured in a hydrogenation reaction of molecule 2, as it is the more stable of the two products. This conclusion can be confirmed by looking at the 3D model of molecule 2: of the two double bonds, the one on the six membered ring is less sterically hindered, and therefore easier to remove. The stability of molecule 4 when compared to molecule 3 can also be highlighted by looking at the table of values above. Molecule 3 has a much higher contribution from bending than the other product, yet has a lower Torsion value (stretching has not been considered due to the relatively low contribution to the total energy). This means that molecule 3 suffers from higher torsional strain - most likely due to the fact that it still contains a double bond in its six-membered ring - and is therefore more unstable. While there are differences in the Van der Waals forces, they are relatively small compared to the bending and torsional contributions, and as such cannot really be considered. This is the same for the H-bonding, which is almost negligible (which is understandable, given that the molecules do not contain any electronegative elements).
Stereochemistry of Nucleophilic additions to a Pyridinium Ring


Molecule 5 (shown in the diagram to the left) reacts with methyl magnesium iodide to form exclusively molecule 6 (also shown in the diagram). The fact that the only product is where the newly added methyl group faces up seems unusual, given that the planar nature of the pyridine ring allows attack from either side. However as the carbonyl group in the molecule directs the Grignard reagent to the same face (see diagram), the carbonyl group must be orientated in a similar way to the new methyl group in the product.
The reactant was modelled in ChemBio3D Ultra and was then geometrically optimised, giving a molecule with the carbonyl group orientated slightly up - this conformation would give molecule 6. However this is not the only possible conformation of the molecule, and the reactant was modelled again after changing its geometry. This was repeated several times in attempts to get new conformations, and after repeated remodelling, three conformers were discovered. These are shown to the left. Despite repeated attempts, no further conformers were made.
-
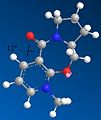 First conformer. Dihedral angle: 12 degrees. Total energy: 180.7 kJ/mol.
First conformer. Dihedral angle: 12 degrees. Total energy: 180.7 kJ/mol. -
 Second conformer. Dihedral angle: 8 degrees. Total energy: 180.8 kJ/mol.
Second conformer. Dihedral angle: 8 degrees. Total energy: 180.8 kJ/mol. -
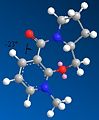 Third conformer. Dihedral angle: -23 degrees. Total energy: 187.7 kJ/mol.
Third conformer. Dihedral angle: -23 degrees. Total energy: 187.7 kJ/mol.



According to the literature[1], there is very low flexibility in the molecule because of its multiple ringed structure. Thus, only two conformational minima are possible: where the carbonyl group sticks up from the molecule, and when it is coplanar to the pyridine ring. This does not agree with the results obtained. However, the first and second conformer have a very similar total energy, and so represent the two conformers listed in the literature. The third conformer, that has a dihedral angle of -23 degrees, does not agree with the literature at all, especially since the literature clearly states that there is no conformation available where the carbonyl group's dihedral angle is below 0 degrees (that is, facing down), due to the nature of molecule 5. However, looking at the third conformer, it is clear that the 5-membered ring containing nitrogen has excessive torsional strain. This shows the limitations to this simple model, as although all elements are correctly angled (i.e. all the carbons in the molecule are roughly tetrahedral) the calculations are only mechanically-based. In order for the calculations to be more accurate, they must take into account molecular orbitals as well. As the model being used doesn't do this, the third conformer - which, as stated above, has a very highly strained 5-membered ring - can be discounted, as it is highly unlikely to exist in any large amount. As the third conformer can be discounted, the results obtained are therefore in agreement with the literature.

The second pyridinium ring structure investigated, labelled as molecule 7, is shown above. This reacts to form the stereospecific product, molecule 8. Once again, the reasoning for this is due to the directing nature of the carbonyl group (as with molecules 5 and 6), and the reactant was therefore modelled in ChemBio3D Ultra to determine the energies of the molecule when the carbonyl group is at different dihedral angles in the molecule.
Several conformers were made and geometrically optimised, and these are shown below.
-
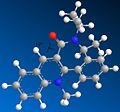 First conformer. Dihedral angle: 19 degrees. Total energy: 277.7 kJ/mol
First conformer. Dihedral angle: 19 degrees. Total energy: 277.7 kJ/mol -
 Second conformer. Dihedral angle: 19 degrees. Total energy: 278.2 kJ/mol
Second conformer. Dihedral angle: 19 degrees. Total energy: 278.2 kJ/mol -
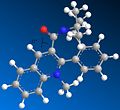 Third conformer. Dihedral angle: -15 degrees. Total energy: 958.5 kJ/mol
Third conformer. Dihedral angle: -15 degrees. Total energy: 958.5 kJ/mol -
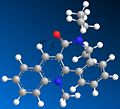 Four conformer. Dihedral angle: 18 degrees. Total energy: 266.1 kJ/mol
Four conformer. Dihedral angle: 18 degrees. Total energy: 266.1 kJ/mol

.jpg)


As shown by the results above, it is clear that three of the four conformers are very similar, displaying similar dihedral angles and energies (although the fourth conformer has the lowest energy - and therefore the most stable - meaning that it is likely to be the most favoured conformation). These conformers all have the carbonyl group facing up, and would all therefore give the proposed product, molecule 8. The only conformation with the carbonyl group facing down has a total energy far higher (over three times larger) than the other conformations, making it highly unlikely to exist in large amounts. Looking at the 3D model, it is clear to see why it has such a high energy - the nitrogen group near the carbonyl suffers from high steric overcrowding.
It can be concluded therefore that the product would have the NHPhenyl group facing up, which means molecule 8 is the principle product. This is in full agreement with the theory.
Stereochemistry and Reactivity of an Intermediate in the Synthesis of Taxol

An intermediate in the synthesis of the molecule Taxol exists as two different atropisomers - with the carbonyl group pointing up (molecule 9) or pointing down (molecule 10).
In order to help determine which of the two isomers are more stable, they were modelled and geometrically optimised on ChemBio3D Ultra. However, the molecule's multiple ring structure and high chance for steric overcrowding (particularly with the two methyl groups on the five-membered ring) means that many varied conformations exist. Most vary by the cyclohexane ring's particular conformation: boat, chair or twist-boat. The chair-based conformation is the most stable of the three (with the boat conformation being the least stable), and a conformation of the intermediate containing the cyclohexane ring in the chair conformation is likely to be the most stable. However, this might not be possible, given its highly strained structure.
Several conformations that were modelled and optimised are shown below.
-
 Carbonyl facing up, boat conformation. Total energy: 276.5 kJ/mol.
Carbonyl facing up, boat conformation. Total energy: 276.5 kJ/mol. -
 Carbonyl facing down, boat conformation. Total energy: 229.1 kJ/mol.
Carbonyl facing down, boat conformation. Total energy: 229.1 kJ/mol. -
 Carbonyl facing up, twist-boat conformation. Total energy: 227.7 kJ/mol.
Carbonyl facing up, twist-boat conformation. Total energy: 227.7 kJ/mol. -
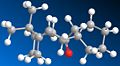 Carbonyl facing down, twist-boat conformation. Total energy: 209.0 kJ/mol.
Carbonyl facing down, twist-boat conformation. Total energy: 209.0 kJ/mol.




Several conclusions can be drawn from the results above. From the total energy values, it is clear that the intermediate's conformation is more stable when the carbonyl group sticks down. The results also show that, as suspected, the boat conformation is the least stable when the carbonyl is sticking either up or down. The twist-boat conformation is more stable, but the most stable conformation must therefore have the carbonyl sticking down and the cyclohexane ring in the chair conformation. This prediction is shown to be true, as it gives an energy value of 204.4 kJ/mol.

When the intermediate's alkene bond is functionalised, the reaction is very slow. This is because it is an example of a hyperstable alkene. According to the literature[2]hyperstable alkenes are slow to react to hydrogenation (and other alkene functionalising reactions) because the loss of the alkene bond raises the molecule's strain energy, and therefore makes it more unstable. This is due to the larger 120 degree angle that a alkene bond allows compared to the smaller 109 degree angle of a tetrahedral carbon. The alkene bond allows molecules with large rings, such as this intermediate, to reach more stable conformations that a single bond would not accommodate. Functionalisation of the double bond is therefore very slow.
Regioselective Addition of Dichlorocarbene

In previous computational experiments, MM2 calculations from the ChemBio3D Ultra program had been used to geometrically optimise and compare molecules. In this experiment, in which the aim was to understand the nature of bonding in dichlorocarbene, the MOPAC/PM6 calculation was also used. This is because results from the MOPAC/PM6 MO calculation method can be used to show the HOMO and LUMO (as well as other) orbitals in molecules, which are used here due to their importance in understanding dichlorocarbene and its reactivity.
Once molecule 11 was modelled in ChemBio3D, it was geometrically optimised using the MM2 calculation. After this, the MOPAC/PM6 calculation was run, and the molecular orbitals were obtained. These are shown below.

-
 LUMO.
LUMO. -
 HOMO - 1.
HOMO - 1. -
 LUMO + 1.
LUMO + 1. -
 LUMO + 2.
LUMO + 2.




Any differences in the two double bonds (which are caused by the chlorine and hydrogen atoms that are bonded to the bridging carbon) in dichlorocarbene can be discovered by looking at the orbitals, specifically the HOMO orbital. Comparing it to the HOMO - 1 and LUMO orbitals, a clear difference can be seen in the two double bonds. The orbitals of the double bond below the Chlorine atom are larger, which means that the double bond below the chlorine is more electrophilic, and therefore more susceptible to nucleophilic attack.
Both molecules were then run through the Gaussian program using B3LYP/6-31G(d,p) calculations. This gave an IR spectra for each molecule, as well as all stretching frequencies. The main stretching frequencies for the two molecules' Cl-C and C=C bonds are shown in the table below.

These results conform the findings from the orbital analysis done earlier in this experiment. Molecule 12 has only one double bond, and this therefore means that the orbitals in molecule 12's Cl-C bond are less crowded (molecule 11's Cl-C bond is destabilised by the second double bond's orbitals). This means that the Cl-C bond is more stabilised and is therefore shorter and stronger. This is reflected in the IR results, which show that the Cl-C bond in molecule 12 has a larger stretching frequency, meaning it is stronger than the Cl-C bond in molecule 11. The fact that the Cl-C bond is shorter also allows the orbitals from the remaining double bond to be stabilised as well, which makes the double bond stronger. This is confirmed by the IR results.
Structure based Mini-project using DFT-based Molecular Orbital Methods
General reaction and the nature of the structure's formation

A paper[3] published in 2005 in the Journal of Organic Chemistry details the controlled synthesis of cis and trans isomers of tetraisoquinoline based compounds. A reaction scheme, shown to the left, was taken from the literature and studied using computational molecular orbital methods.
The more polar intermediate shown in the reaction scheme is, as described in the literature, formed rapidly by isomerisation from the reactant once the PdCl2(PhCN)2 in THF has been added. Although some of the two isoquinoline isomers are formed directly from the first reactant, most is formed from the intermediate.
The product that the reaction forms is almost exclusively the cis isomer - the ratio of the cis-isomer to the trans-isomer is 10:1. There are several possible reasons for this. One reason for the cis-isomer's dominance may be due to the molecular orbitals of the two different isomers.
-
 HOMO orbitals of the trans-isomer.
HOMO orbitals of the trans-isomer. -
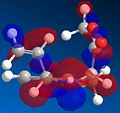 HOMO orbitals of the cis-isomer.
HOMO orbitals of the cis-isomer.


As the HOMO orbitals above show, the cis-isomer has a larger area of orbitals in its HOMO than the trans-isomer does, increasing stability. Another key difference is that the orbitals around the nitrogen atom on the cis-isomer fill a greater than on the trans isomer, which means that they are less crowded and are therefore more stable. Although the position of the alkene group is the only difference between the two isomers, it has a large effect, and by looking at the HOMOs of the two molecules it seems likely that the cis-isomer would be favoured over the trans-isomer.
Another reason for the cis-isomer's near exclusive formation may be due to the nature of the reaction. It is stated in the literature that the the Pd(II) used in the reaction, which induces the cyclisation and causes the formation of the tetrahydroisoquinoline, bonds to part of the molecule in the transition state and 'guides' the product to a particular conformation - in this case the cis-isomer.

Spectroscopic analysis of the molecule
The 13C NMR spectra of both geometrically optimised isomers was calculated using Gaussian. The data is shown below.
-
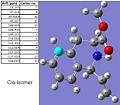 Gaussian generated 13C NMR data of the cis-isomer.
Gaussian generated 13C NMR data of the cis-isomer. -
 Gaussian generated 13C NMR spectrum of the cis-isomer.
Gaussian generated 13C NMR spectrum of the cis-isomer. -
 Literature 13C NMR spectrum of the cis-isomer.
Literature 13C NMR spectrum of the cis-isomer.



-
 Gaussian generated13C NMR data of the trans-isomer.
Gaussian generated13C NMR data of the trans-isomer. -
 Gaussian generated 13C NMR spectrum of the trans-isomer.
Gaussian generated 13C NMR spectrum of the trans-isomer. -
 Literature 13C NMR spectrum of the trans-isomer.
Literature 13C NMR spectrum of the trans-isomer.



Although the 13C NMR spectra generated by Gaussian look fairly similar to the literature spectra, when the chemical shift values are compared, there are vast differences. The Gaussian generated spectra also do not take into account 13C - 13C coupling, meaning that, although there is a large and messy multiplet at around 128ppm in the literature, the Gaussian generated 13C spectra does not have it. This means that the Gaussian program was ineffective at calculating this sort of spectra, apart from to a very rough degree of accuracy.
However, the 13C spectra of the two isomers are virtually identical, which means that would not be used to tell them apart in a lab anyway. Although the literature does not supply any IR spectra, IR spectra of the two isomers were created using Gaussian in order to see if they were different enough for the isomers to be identified seperately if they were synthesised in a lab. Although both of the IRs are very similar, there is a key difference: the trans isomer, due to the nature of the alkene, has more space for the carbonyl bond to vibrate (the cis-isomer's carbonyl bond is more hindered). This gives the trans isomer a larger carbonyl peak (at around 1750cm-1) than the cis-isomer. Therefore, if the two isomers were synthesised and needed to be identified, IR would be a more effective way of telling them apart than 13C NMR.
-
 Gaussian generated IR spectrum for the trans-isomer.
Gaussian generated IR spectrum for the trans-isomer. -
 Gaussian generated IR spectrum for the cis-isomer.
Gaussian generated IR spectrum for the cis-isomer.

