
Organic:luke4912
Computational Labs - Module 1: Structure and Spectroscopy
The Hydrogenation of Cyclopentadiene Dimer
Dimerisation


Cyclopentadiene undergoes facile dimerisation to either its endo or exo dimer product, shown in Figure 1. To deduce which product has the lower energy conformation (and therefore is the preferred isomer), both molecules were modelled using ChemBio3D Ultra software and their energies minimised using the MM2 geometry optimisation method.
The computed energy values are meaningless on their own, but by relative comparison it was seen that product 2, the exo isomer was the preferred dimer having a lower energy of 133.496 kJ/Mol. The endo product had an energy of 142.393 kJ/Mol. The exo isomer is the thermodynamic product and therefore if the endo isomer is produced, the reaction must be kinetically controlled. Sterics can explain these observations: in the endo isomer the bridging methyl group is hindered by the two hydrogens also sticking up from the ring - this would cause extra strain within the molecule, raising its energy. This can be seen in Figure 2.
Hydrogenation
The endo isomer is able to undergo monohydrogenation at either alkene within its structure, these are shown as products 3 and 4 in Figure 1. The above procedure was employed in the same manner to deduce what the most likely product of this reaction is. Table 1 shows a breakdown of the energies for each product, along with the total. It can be seen that product 4 is the lower energy conformer and thus the favoured product of monohydrogenation. This is also confirmed in the literature[1].

It is the alkene of the 6-membered ring that is hydrogenated, again this is due to steric reasoning. The alkene of the 5-membered ring is more hindered and thus harder to attack. Table 1 shows that product 3 has significantly more bending strain than product 4, due to the crowded 5-ring having to spread its extra hydrogens apart. However, product 4 does have more torsional strain - arising from 5-ring alkenes having smaller bond angles than 6-ring. All other energies are approximately matched.
Stereochemistry of Nucleophilic Additions to Pyridinium Ring


In the reaction scheme shown in Figure 3 the methyl group is delivered with absolute stereochemistry to form the product 2. Initially this seems strange because the planar nature of the pyridium ring would suggest attack is possible from either side to give a racemic product mixture. When modelling and optimising the reactant, as previously described, it was found that in the lowest energy conformer (180.537 kJ/Mol) the amide carbonyl sits above the pyridinium ring with a dihedral angle of 24 degrees (shown in Figure 5).
This explains the stereochemistry of the product - when the Grignard's Mg atom chelates to the carbonyl oxygen it has no choice but to deliver the methyl group to the top side of the ring. (Shown in Figure 4.) Upon experimentation with the model it turns out there are two conformational minima, the aforementioned carbonyl-above-the-ring isomer and also a coplanar isomer - here the carbonyl is approximately inline with the pyridinium ring, but it still has a slight dihedral angle of 12 degrees. This is also seen in Figure 5.
These results are attributed to the inherent inflexibility of the 7-membered ring in the molecule[2], there is no energetically plausible way have the carbonyl sitting below the ring, it would need the rest of the entire molecule to distort. This model fails to take into account the molecular orbitals of this molecule because it is purely a mechanical approach to modelling, this may mean some elements are missed from the calculations.
-
 Figure 4: CO above ring (dihedral angle: 24 degrees) Energy: 180.537 kJ/mol.
Figure 4: CO above ring (dihedral angle: 24 degrees) Energy: 180.537 kJ/mol. -
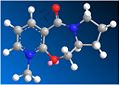 CO coplanar (dihedral angle: 12 degrees) Energy: 187.142 kJ/mol.
CO coplanar (dihedral angle: 12 degrees) Energy: 187.142 kJ/mol.


The second pyridinium system, shown in Figure 6, reacts with very different stereochemistry. Once modelled and optimised, the lowest energy conformation of the reactant shows the amide's carbonyl to be sitting below the plane of the pyridinium ring, with a dihedral angle of ~-20 degrees. Figure 7 shows an experimentation of carbonyl orientations, all of which are below the ring after optimisation.
The unexpected stereochemistry of the product (the reverse of the first reaction) is due to the stability inferred by dipolar coupling[3] present in that orientation of the molecule. The methyl group sticking up from the ring couples with the alpha phenyl hydrogens. This indicates the amine attacked on the opposite face of the carbonyl, hence the diastereoselectivity seen.

-
 Figure 7: Dihedral angle: -19 degrees. Energy: 265.859 kJ/mol.
Figure 7: Dihedral angle: -19 degrees. Energy: 265.859 kJ/mol. -
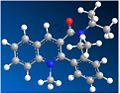 Dihedral angle: -20 degrees. Energy: 289.080 kJ/mol.
Dihedral angle: -20 degrees. Energy: 289.080 kJ/mol. -
 Dihedral angle: -22 degrees. Energy: 278.176 kJ/mol.
Dihedral angle: -22 degrees. Energy: 278.176 kJ/mol.



Stereochemistry and Reactivity of an Intermediate in the Synthesis of Taxol

The two possible atropisomers of an intermediate in Taxol synthesis are shown in Figure 8. To determine the more favourable form (carbonyl pointing up or down), both molecules were modelled and optimised as previously discussed. The results obtained were very interesting. Due to the multiple ring structure of these molecules there were lots of different optimisation products: the carbonyl orientation was accompanied by changes in the cyclohexane ring, from its chair to twist-boat to boat conformers.
Figure 9 shows the most prominent conformers with their energies. Knowing cyclohexane's lowest energy form is its chair conformation, the favoured Taxol product should display a chair orientation.
-
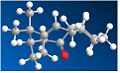 Figure 9: CO down, chair cyclohexane - Energy: 185.424 kJ/mol.
Figure 9: CO down, chair cyclohexane - Energy: 185.424 kJ/mol. -
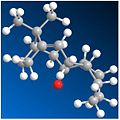 CO down, twistboat cyclohexane - Energy: 209.994 kJ/mol.
CO down, twistboat cyclohexane - Energy: 209.994 kJ/mol. -
 CO up, chair cyclohexane - Energy: 505.137 kJ/mol.
CO up, chair cyclohexane - Energy: 505.137 kJ/mol. -
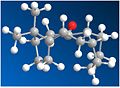 CO up, twistboat cyclohexane - Energy: 530.593 kJ/mol.
CO up, twistboat cyclohexane - Energy: 530.593 kJ/mol.




The results show, for orientations with the carbonyl pointing up the molecule's energy is very high and these conformers are unlikely to form. Where the carbonyl points down, the chair conformer is indeed the lowest energy molecule, followed by the twist-boat arrangement (with a difference of ~24 kJ/Mol) and the boat arrangement would simply not form upon experimentation - suggesting it would be very unfavourable.
According the literature[4] a hyperstable alkene is slow to react to hydrogenation because of the alkene element (thus carbon hybridisation) would raise the molecules strain and torsion energies. A alkene, sp2 bond is more flexible, has a wider bond angle and can rotate more than a 4-centre sp3 carbon bond. Increasing strain obviously increases instability and thus molecular energy.
Regioselective Addition of Dichlorocarbene
To understand the bonding in dichlorocarbene (Figure 10), a MOPAC/PM6 calculation was performed in addition to the standard MM2 optimisation used throughout this report. This allowed molecular orbitals to be plotted, from which the reactive behaviour of the molecule could be explored.
Key orbitals of the molecule are shown below in Figure 11. The chlorine of the bridging carbon group has a clear affect on the molecule, because the molecular orbitals of the two alkene groups are quite different. The HOMO diagram shows the alkene underneath the chlorine has a much larger negative molecular orbital and thus electron density so it is much more susceptibile to electrophilic attack. This double bond would be the first to react to an incoming electrophile. Both molecules in Figure 10 were then subject to Gaussian calculations (B3LYP/6-31G(d,p)) in order to retrieve infrared spectra. The most prominent signals are shown below in Table 2, these are the Cl-C & C=C bond responses.
-
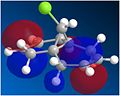 Figure 11: The LUMO+2 orbital
Figure 11: The LUMO+2 orbital -
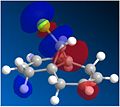 The LUMO+1 orbital
The LUMO+1 orbital -
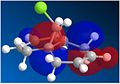 The LUMO orbital
The LUMO orbital -
 The HOMO orbital
The HOMO orbital -
 The HOMO-1 orbital
The HOMO-1 orbital





{kind=link}
The results show a stronger Cl-C bond in the single alkene species. The bond suffers no crowding or destabilisation from the second alkene group. There is also a strengthening of the C=C bond, due to the stronger C-Cl (ie. shorter bond) meaning the C=C bond's molecular orbitals are more stabilised - more space.

Mini-Project - Stereoselective dissolving metal reductions

In the above reaction, the ketone reactant is reduced with stereochemical control to give almost exclusively the alcohol shown, where the -OH group points down. This mini-project will confirm this is indeed the correct product. By modelling the product and its isomer (with the -OH group pointing up) and optimising them using MM2/MOPAC methodology, they were then put through Gaussian in order to obtain relevant spectroscopic information.

The labelled model of the product above corresponds to the calculated NMR values in the table below. About half the carbon signals dont match the literature values, suggesting the 13C spectra needs reassigning. You would be able to tell this reaction had gone to completion from its IR spectrum, there would be a disappearance of the keto- C=O bond at ~1600cm-1 and a new signal at ~3900cm-1 to indicate the presence of an -OH bond. To determine the correct isomer, optical rotations of the product(s) would need to be determined, these are also shown in the table below. These values, complete with calculated J3 values (obtained from using the Karplus equation), show that general good agreement between the literature values and the gaussian -OH down values mean the main product must be the one where the -OH points down.

References
[1] - A. Francisco, Tet Lett, 1996, 37, 6925-6928
[2] - A. G. Shultz, L. Flood and J. P. Springer, J. Org. Chemistry, 1986, 51, 838
[3] - Leleu,S et al., Tetrahedron: Asymmetry, 2004, 15, 3919-3928
[4] - S.W Elmore et al, Tet Lett, 1991, 319