
Namespace:jr407

Computational Chemistry - Module 1: Organic
Modelling Techniques
The computational models described below are built from structural drawings (ChemDraw) which have been converted to 3D structures using Chem3D (from where 2D [.jpg] and 3D JMol visuals [.mol]) have been produced. Structural optimisations (MM2) calculations are run to find lowest energy conformers (Chem3D), this a molecular mechanics based approach.
Molecular mechanics is limited as it operates from defined parameters for bond lengths and angles, accounting only for basic lone pair interactions, sterics and van der Waals forces - there is no account for molecular orbitals and secondary orbital interactions. This often results in local energy minima structures being given, as alteration is only within a limited range of the starting structure. Consideration of where electrons locate in molecules is essential for predicting stereoselectivity in certain reactions, such as Diels Alder cycloadditions. More advanced (and time consuming!) semi-empirical molecular orbital theory takes these factors into account, giving a more reliable result.
Key literature
- H. O. House, J. L. Haack, W. C. McDaniel, and D. VanDerveer, Enones with strained double bonds. 8. The bicyclo[3.2.1]octane system, J. Org. Chem., 1983, 1643-1654. DOI: DOI:10.1021/jo00158a014 , DOI:10.1021/jo00158a014
The Hydrogenation of cyclopentadiene dimer

Cyclopentadiene dimerises to produce specifically the endo dimer 2 rather than the exo dimer 1, despite the higher energy of 2: the exo dimer is less strained / hindered being thermodynamically favourable, thus the reaction is under kinetic control (the endo dimer has a lower energy more stable transition state, as a result of greater p orbital overlap). Hydrogenation of this endo dimer proceeds to give initially one dihydro derivatives (4); the energies of the four molecules are tabulated below (MM2 Chem3D optimisation), and their structures shown (right - click buttons for JMol 3D visuals).

| Relative Energies of the Cyclopentadiene Dimers & Dihydro Derivatives | Thermodynamic Vs. Kinetic Control - Illustration | |||
|---|---|---|---|---|
| Molecule | Energy / kcal/mol | Comment | Conformer |  |
| 1 | 31.8782 | Minor Product | Exo | |
| 2 | 34.0030 | Major Product | Endo | |
| 3 | 35.9334 | Minor Product | LHS Double Bond Remains | |
| 4 | 31.1587 | Major Product | RHS Double Bond Remains | |
|
Hydrogenation of the cyclopentadiene dimer | ||||
The higher energy of the endo dimer 2 is explained by the increases steric hindrance and Van der Waals forces as the structure bends round, being less planar.
The relative contributions from the stretching (3 ~ 4), bending (3 > 4),torsion (3 ~ 4), van der Waals (vdw 1,4: 3 > 4) and hydrogen bonding (H-bond: 3 ~ 4) energy show 4 to be more stable than 3; in all cases: bending > torsion > stretching > vdw > H-bond. The principal difference however arises from bending energy, which is significantly greater for 3, as a result of increased steric hindrance. The MM2 (Chem3D) energy minimising optimisation data on each energy of 3 and 4 is tabulated below.

| Energy Type | 3 Energy / kcal/mol | 4 Energy / kcal/mol |
|---|---|---|
| Stretch | 1.2211 | 1.0979 |
| Bend | 18.8397 | 14.5569 |
| Torsion | 12.2563 | 12.5052 |
| Van der Waals (1,4) | 5.7632 | 4.5052 |
| Dipole - Dipole | 0.1631 | 0.1406 |
Thus the thermodynamic prediction (lowest energy) is that product 4 is the major. This may also be rationalised by ring strain - the double bonded sp2 carbons yearn for a bond angle of 120, the greater the deviation from this the more strained the system. The LHS (left hand side) double bonded carbons have a C-C-C bond angle of 108 in comparison to the RHS (right hand side) which is 113, thus hydrogenation of the LHS double bond gives the greatest reduction in ring strain (producing tetrahedral sp3 carbons which typically have 109 bond angles). Thus product 4 dominates, with the double bond retained on the RHS and hydrogenated on the LHS.
Stereochemistry of Nucleophilic additions to a pyridinium ring (NAD+ analogue).





Two reactions are shown below - the first involves the optically active derivative of prolinol (5) which reacts with methyl magnesium iodide to alkylate the pyridine ring in the 4-position, with the absolute stereochemistry shown in 6. In the second example, the pyridinium ring of 7 is derivatised by reaction with aniline to form 8 which acts to transfer the NHPhenyl group to electrophiles (e.g. carbonyl groups) in a reverse of the reaction by which it was formed. The stereochemistry of 8 is most likely a result of passing through a lower energy endo transition state, thus giving an endo product (as discussed in the example above). The reagent and product structures are shown right, with JMol 3D visuals for 5 and 7 viewable by clicking the buttons below.
The (structurally optimised) energy of 5 is 43.1197 kcal/mol (which decreases to 37.4754 kcal/mol upon formation of product 6). The energy of 7 is 63.4021 kcal/mol (decreasing to 58.8804 kcal/mol upon formation of product 8).
Prolinol Derivative 5
Notably the MeMgI molecule cannot be modelled in Chem3D as the Mg element is not supported (no data in the programme for it).
Varying the geometry of the carbonyl group and its orientation with respect to the aromatic ring always yields the same result, proving this to be the optimal - orientating the CO bond above and below the aromatic ring are shown to the right. Similarly the disubstituted carbon in the same ring may be orientated above or below the ring - optimisation again yielding the same nearer to in plane result (dihedral angle of ~10 between CO and ring).
A distinct drawback of the molecular mechanics approach is it only resolves and optimises within a certain range of the intital input - thus if the molecule is distorted enough, then the optimisation will give a different higher energy conformer. This merely illustrates a limitation of this modelling approach. Such a conformer is shown right, which has an optimised energy of 145.4803 kcal/mol.
In reaction of 5 with MeMgI, the Mg atom coordinates with the carbonyl O, as the Me is delivered to the aromatic ring, via a 6 membered ring in the transition state. As shown (right, JMol visuals) the carbonyl is angled just slightly above the ring, thus the Me group is delivered to the top side of the ring - this mechanism is shown below.
 |
 |
 |
|
Pyridinium Derivative 7
Similar to the prolinol derivative (5) the carbonyl group in the pyridinium derivative (7) is also angled slightly above the planar aromatic rings (by ~20). The difference here though is that in the reaction to product 8 the attacking nucleophile (via the lone pairs of the nitrogen) attacks from the opposite face (avoiding the electron rich C=O oxygen), thus giving a stereochemistry where the secondary amine group and methyl are on the same face of the molecule (opposite to the carbonyl), this can be seen in the JMol 3D visual above, where the mechanisms and carbonyl angle in 7 are also shown.
Limitations of Molecular Mechanics
The molecular mechanics modelling technique is effective but limited - as illustrated above - taking into account only basic lone pair repulsions, sterics / van der Waals forces and supplied data on bond lengths and angles: there is no account of molecular orbital (MO) interactions, for example mixing may occur significantly altering the outcome.
Key literature
- A. G. Shultz, L. Flood and J. P. Springer, J. Org. Chemistry, 1986, 51, 838. DOI:10.1021/jo00356a016
- Leleu, Stephane; Papamicael, Cyril; Marsais, Francis; Dupas, Georges; Levacher, Vincent. Tetrahedron: Asymmetry, 2004, 15, 3919-3928. DOI:10.1016/j.tetasy.2004.11.004
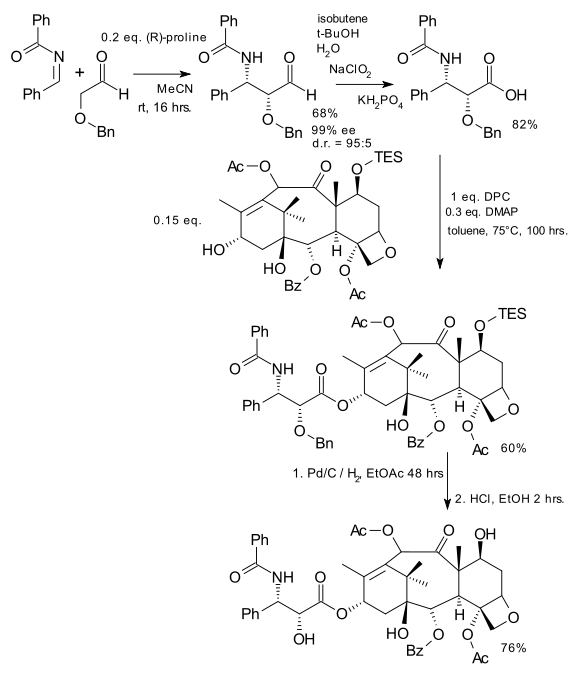
Stereochemistry and Reactivity of an Intermediate in the Synthesis of Taxol

The total synthesis of Taxol involves an intermediate with a carbonyl group pointing either up (9 - a twist-boat conformer) or down (10 - a chair conformer). This intermediate then isomerises to the alternative carbonyl isomer (atropisomer). Isomer 10 is the more stable with a structurally optimised (MM2) energy of 44.3000 kcal/mol, in comparison to 9 at 59.6757 kcal/mol; notably upon running optimisations both rearrange to give 10, 9 only being achieved by manual distortion of the molecule and optimisation to give a local minima in energy. The higher energy of 9 is a result of the upward carbonyl corresponding to the cyclohexane and cyclopentane rings pointing downwards and generating steric hindrance, which is not present in 10. It is difficult to lower the energy any further by manual distortion and optimisation, with this only leading to higher energy conformers, for example 11 - viewable as a JMol 3D by clicking right - at an energy of 122.7725 kcal/mol, with the cyclohexane ring now in a twist-boat confirmation, as opposed to the chair form present in isomers 9 and 10.
The energy of 10 arises from 1,4 van der Waals forces, bending and torsion. This arises in part from a dihedral angle of 42 between the carbonly oxygen and hydrogen on the neighbouring disubstituted carbon, as opposed to the optimum staggered angle of 60. Bending energy in part arises from the cyclopentane ring straining the double bonded sp2 carbon to a 108 C-C-C bond angle, as opposed to the desired 120. Steric hindrance also arises between the hydrogens of the methyl groups on the bridging carbon and the hydrogens of the decane ring.
The alkene reacts particularly slowly as it is a hyperstable alkene[1] - that is the alkene gives the molecule a lower energy than the alkane equivalent would. This hyperstability may be overcome by increasing the driving force of the reaction. An example is the use of m-chloroperbenzoic acid (CHCI,, NaOAc, 20°C) to oxidise such alkenes to epoxides[2].
From intermediate 10 the synthesis continues to deliver hydoxyl groups to the top face (pointing up, as opposed to the carbonyl group which is pointing down) of the molecule, and an OBz functional group to the bottom face (opposite side of ring to carbonyl group), as part of the final taxol product. This can be seen in the synthetic route here: http://chem.vander-lingen.nl/uploads/taxol_sidechainadditiondziedzic_2009.svg.png.
{kind=link}
Key literature
- S. W. Elmore and L. Paquette, Tetrahedron Letters, 1991, 319; DOI:10.1016/S0040-4039(00)92617-0 10.1016/S0040-4039(00)92617-0 10.1016/S0040-4039(00)92617-0
- J. G. Vinter and H. M. R. Hoffman, J. Am. Chem. Soc., 1974, 96, 5466; DOI:10.1021/ja00824a025 DOI:10.1021/ja00824a025 & 95
- Vancomycin, J. Am. Chem. Soc., 1999, 121, 3226; DOI: 10.1021/ja990189i
Regioselective Addition of Dichlorocarbene


Compound 12 - shown right - has reactivity dictated by orbital control: here this is illusrated using the electrophile dichlorcarbene in example. The molecule's structure has been optimised (MM2) in the classical molecular mechanics method, then further with MOPAC/PM6, being a quantum approach in so far as giving an approximate wave description of the valence electrons. The principal molecular orbitals for determining reactiviy are shown below: this has also been repeated for compound 13 - hydrogenation of the anti / exo double bond in 12. The initital MM2 calculation found molecule 12 to have an energy of 17.8972 kcal/mol, and 13 an energy of 29.2172 kcal/mol.




Visual inspection of the MO diagrams shows a large electron density cloud in the HOMO of the dialkene (12) between the C=C bond and Cl-C bond (left hand side of molecule): this results from the electronegative Cl atom drawing electron density from the electron rich alkene bond. Such a feature is clearly not present in the monoalkene (hydrogenation product 13), neither is it present on the right hand side of molecule 12. It is this large electron cloud that makes the endo alkene more nucleophilic and reactive.
Notably a molecular mechanics calculation would not distinguish the two alkenes, but the semi-empirical method used here accounts for molecular orbitals and thus does distinguish, quantifying the effect of the Cl on the different alkenes - this is shown in vibrational energy.
The molecules' vibrations are found (density functional approach) from the optimised structure, further optimising with Gauss and then running a Gaussian calculation, producing the infrared spectra shown right. The C-Cl and C=C vibrations are then found: this has been repeated for variation of the alkene substituents from electron withdrawing groups (F) to electron donating groups (CO), via OH (and H, being 12 and 13). Typically one would expect an electron withdrawing group to weaken a bond decreasing the energy required to make it vibrate and thus decreasing the frequency of absorption (by E = hv) - the results are tabulated below (right) with the structures of the molecules studied (below, left) and discussed.
For compound 12 (dialkene) the principal C-Cl absorption is at 770.91, which is in agreement with the literature[3]; notably many of the vibrations result in some peturbation of the bond, but to a lesser extent. The C=C absorptions are at 1757.37 (on the Cl side) and 1737.11, both being intense stretches of the bond. For the hydrogenation product (monoalkene - compound 13) the C-Cl absorptions are in a similar range; the single C=C peak occurs at 1736.73, being near identical to that in the dialkene case.

The IR spectra of each of the six compounds with varied substituents can be viewed by the following links:
- CO Substituted Dialkene
- CO Substituted Hydogenated (Monoalkene)
- OH Substituted Dialkene
- OH Substituted Hydogenated (Monoalkene)
- F Substituted Dialkene
- F Substituted Hydogenated (Monoalkene)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
From the graph it can be seen that the more likely the substituent is to donate electron density, the stronger the C=C bond, and thus the more energy it takes to make it vibrate (E = hv) and so the higher the frequency of IR absorption. This is rationalised thus: the carbonyl group is electron withdrawing, the hydroxyl is mildly electron donating, via the lone pairs on the oxygen atom into the π * orbitals of the C=C bond; finally - and most interestingly - the F atoms are so electronegative and electron withdrawing that they have an effect through the molecule, pulling the two carbons closer together and shortening the C=C bond (effectively increasing the density) strengthening it.[4]
Key literature
B. Halton, R. Boese and H. S. Rzepa., J. Chem. Soc., Perkin Trans 2, 1992, 447. DOI:10.1039/P29920000447
Structure Based Mini-Project

Here the selectivity of a napthoquinone derivative forming reaction is discussed, using computational methods to predict spectra and comparing these with the literature, to conclude on whether the major product (of two isomers) may be determined computationally. The product compound exhibits antiprotozoal (which may cause some of the most clinically severe lessions in tropical diseases) activity with lower cytotoxicities than existing molecules. The findings are reported by Tapia et al.[5]
- Introduction to reaction and isomers
Two isomers (10A and 10B) structures are shown left, with a JMol 3D visual of 10B: this doesn't show a distinct C=N double bond, as there is infact delocalisation across N-C-S and up to the aromatic ring. The reaction for their formation is shown below.

The reaction is reported in the literature, where a mixture of the two isomers was formed in the ratio A:B of 87:13. The principal characteristic that distinguishes the isomers is melting point: 243-244C for A, and 236-237C for B; the isomers are physically separable by column chromatography (silica gel with dichloromethane/petroleum ether [5/95]).
The products 10A and 10B (thiazole o-quinodimethanes: o-QDMs) are formed through a Diels–Alder trapping of a naphthoquinone 2, followed by deprotonation to give the aromatic ring shown. Interestingly the stereochemistry is reversed by addition of Br to the 6 position of 2 (N.B. position 1 is the dimethyl, numbering clockwise).
The experimental procedure is thus: a solution of 4-(bromomethyl)-5-(dibromomethyl)thiazole (0.216 g, 0.6 mmol) in dry DMF (2.0 mL) was slowly added to a stirred and heated solution, at 60 &C, of the quinone '2' (0.5 mmol) and NaI (5 equiv) in DMF (3 mL). Stirring and heating were maintained for 1h. After cooling, the precipitate was filtered off and washed with water and then with ethyl acetate. Hence the proposed mechanism of base giving deprotonation; the final stage of the mechanism is oxidation of the cyclohexadiene, to eliminate two hydrogens: this may run by a radical mechanism and / or involve the Br- and Na+ ions.
- Minimisation and energy
Initial MM2 geometry optimisation gives an energy of 29.8190 kcal/mol for 10B and 29.7908 kcal/mol for 10A. Further optimisation was performed by MOPAC/PM6 calculations and Gaussian optimisation (DFT=mpw1pw91; basis set: 6-31G(d,p)). These results show isomer 10A to be marginally more stable, and thermodynamically the predictable dominant isomer.
- 13C NMR prediction (both isomers) and comparison to literature
The prediction was run in Gaussian (again DFT=mpw1pw91; basis set: 6-31G(d,p)), the predicted spectra of the two isomers are shown right (click to enlarge). The calculation jobs can be found in the digital repository (D space) here: 10B: DOI:10042/to-2915 ; 10A: DOI:10042/to-2941 .


Using the labelled model of 10A (shown right - click to enlarge) the chemical shifts of the carbon environments for the predictions and literature (A and B) are tabulated below-right (solvent: CDCl3; TMS ref.), these results are then graphed (below left).

Notably the vast majority of the peaks have highly similar values for both isomers, the predicted and literature; expansion of the graph for carbon numbers 3-6 shows the literature values to be typically slightly higher. Expansion at carbons 11 and 12 shows how the isomers may be distinguished: repositioning of the oxygen defines the shift for these carbons, that adjacent to the O having the higher shift.
Furthermore, the carbon adjacent to the ether oxygen and in the aromatic ring has a higher shift for isomer 10A, where the carbon is spatially closer to nitrogen. In contrast the shift for such a carbon is slighlt lower in 10B, when the carbon is now spacially closer to sulphur than nitrogen.



{kind=link}
{kind=link}
- 3J H-H couplings
There are no such couplings present in the molecule so such an analysis cannot be performed.
- Predicting the IR spectrum
ΔG = ΔH - T.ΔS: the free energy difference between the molecules may be found - 10A has a free energy of 788 563.2 kcal/mol (-1256.674465 Hartrees) and 10B a free energy of 788 563.1 kcal/mol - which is negligible.
The predicted IR spectra of each isomer are shown right with links to 3D visuals of two key vibrations: the interesting aromatic ring distortion, and the CO stretch adjacent to the ether O (click to view). The kay vibrations are tabulated and analysed below.

The IR spectra is somewhat limited in determining which isomer is which, with little difference between peaks (~3cm-1) . Furthermore, the CO absorption (near the ether oxygen) is predicted to be higher for 10A, but the literature gives a higher value for 10B; in addition the literature values are typically higher than the predicted (by 5-15cm-1), even accounting for an approximate 8% error in stretching frequencies. Thus practical applications are limited.
On theoretical levekl however, in both isomers the predicted absorptions associated with CH vibrations are the same, as one would expect, as are the absorptions for aromatic ring distortion.
Then there is some difference when one considers the C=N and C-S stretches - these tend to be higher (stronger bonds) when the group is located between the ether O and S: i.e. the C=N stretch is higher for 10A than 10B. A plausible explanation is that the group is more central to the majority of electron density, with an effect dipole present in the molecule (as shown above). Such a theory is supported by the 13C NMR, considering carbon pairs '3 & 6' and '10 & 13' (of 10A - see labelling on NMR spectra) we see a higher shift for the carbon on the bottom / electron poor side of the molecule (that is on the opposite side of the main back bone to the ether O). This stronger C=N bond and theory can be extrapolated to imply that 10A is the more stable product, and so would be the dominant isomer.
- Predicting the optical rotation & circular dichroism
Both isomers are not optically active as they contain neither chiral centre nor planes of chirality (the mirror image of each molecule can easily be superimposed by simple rotation). This said, running an optical rotation calculation in Gauss for isomer 10A returned a value of [Alpha] (5890.0 A) = -2.55 deg, instead of 0 deg as expected. Notably the molecule contains 20 non-hydrogen atoms, making such a calculation very taxing on the system used, and this is an estimate, also being unreliable at such a low value, which explains in part why a precise value of 0 deg does not arise.
- Predicting selectivity by consideration of frontier molecular orbitals (FMOs)
Analysis of FMOs by semi-empirical PM3 (Chem3D) shows the greatest HOMO coefficient to be located on the carbon bearing bromine in reagent 9, this also being rendered partially positively charged by the electronegative Br atom, and on carbon 5 in reagent 2 there is the more partial negative charge than on carbon 6: thus on combination the partial positive and negative charges align to give isomer 10A as the preferred result, and thus the dominant product. This is illustrated below; notably there is little difference in LUMO coefficients between carbons 5 and 6 in 2, contrary to the implication of the literature.

- Conclusion
Computational methods predict the 13C NMR and IR spectra to good accuracy, and imply that isomer 10A should be the dominant product (thermodynamically), which geometry optimising energy calculations confirm also. This is indeed the case according to the literature: this demonstrates the power of computational methods in predicting reaction outcomes, but this is not without its limitations - should a reaction be under kinetic control, the computational prediction may be overturned if orbital considerations are not made or a full quantum calculation(though this is not the case here). In addition, the spectroscopic methods dicussed are not able to distinguish the isomers in the lab (the data for the different isomers are too close, e.g. 3cm-1 in IR), here a chromatographic separation is required and physical analysis.
Key literature
R. A. Tapia et al., Bioorganic & Medicinal Chemistry, 2003, 11, 2175-2182.
References
- ↑ 'Evaluation and prediction of the stability of bridgehead olefins' - Wilhelm F. Maier, Paul Von Rague Schleyer -Journal of the American Chemical Society 1981 103 (8), 1891-1900
- ↑ 'Bissecododeca hedraenes, Unusual Hyperstable Olefins' - Paul R. Spurr, Bulusu A. R. C. Murty, Wolf- Dieter Fessner, Hans Fritz, and Horst Prinzbuch -Angew Clwm In,. Ed. Engl. 1987 26 (5), 455-456
- ↑ Silverstein, R.M.; Bassler, G.C. and Morrill, T.C. Spectrometric Identification of Organic Compounds, 4th ed., New York: John Wiley & Sons, 1981
- ↑ 'Understanding organofluorine chemistry. An introduction to the C–F bond' - D. O'Hagan - Chem Soc Rev 2008 37 (2), 308–319
- ↑ 'Synthesis and Antiprotozoal Activity of Naphthofuranquinones and Naphthothiophenequinones Containing a Fused Thiazole Ring' - R. A. Tapia et al. - Bioorganic & Medicinal Chemistry 2003 (11), 2175-2182
External links
- [Taxol Total Synthesis: http://chem.vander-lingen.nl/uploads/taxol_sidechainadditiondziedzic_2009.svg.png]
- [Chemistry SCAN Link: https://scanweb.cc.imperial.ac.uk/uportal2/]