
Molecular Modelling : Justin Wilson
Molecular Modelling 2
NH3 Molecule
Ammonia |
Calculation Method: RB3LYP
Basis set: 6-31G(d,p)
E(RB3LYP): -56.55776873
Point Group: C3v
N-H Bond Length: 1.01798Å (lit: 1.012Å)1
H-N-H Bond Angle: 105.741
Item Value Threshold Converged?
Maximum Force 0.000004 0.000450 YES
RMS Force 0.000004 0.000300 YES
Maximum Displacement 0.000072 0.001800 YES
RMS Displacement 0.000035 0.001200 YES
Predicted change in Energy=-5.986308D-10
Optimization completed.
-- Stationary point found.
----------------------------
! Optimized Parameters !
! (Angstroms and Degrees) !
-------------------------- --------------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------------------
! R1 R(1,2) 1.018 -DE/DX = 0.0 !
! R2 R(1,3) 1.018 -DE/DX = 0.0 !
! R3 R(1,4) 1.018 -DE/DX = 0.0 !
! A1 A(2,1,3) 105.7412 -DE/DX = 0.0 !
! A2 A(2,1,4) 105.7412 -DE/DX = 0.0 !
! A3 A(3,1,4) 105.7412 -DE/DX = 0.0 !
! D1 D(2,1,4,3) -111.8571 -DE/DX = 0.0 !
--------------------------------------------------------------------------------
{kind=link}
Charge Distribution:
As Nitrogen is significantly more electronegative than Hydrogen, one would expect that the Nitrogen atom will draw electron density from the covalent bond away from the Hydrogen. Therefore the Nitrogen ought to have a large negative charge and the Hydrogens ought to have a smaller, positive charge.
N= -1.125 H= 0.375
Questions + Answers
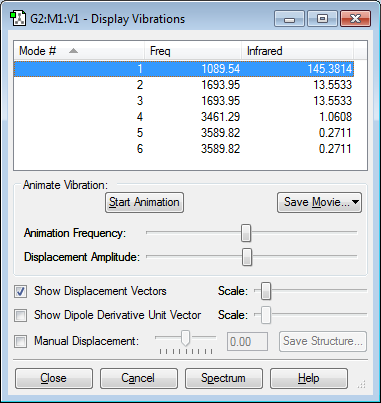
How many modes do you expect from the 3N-6 rule?
In most cases, non-linear molecules with N number of atoms will have 3N-6 normal modes of vibration. Ammonia has 6 normal modes.
Which modes are degenerate (ie have the same energy)?
5,6 and 2,3 are degenerate modes of vibration as they have the same energy.
Which modes are "bending" vibrations and which are "bond stretch" vibrations?
1,2,3 are bending vibrations as the Hydrogen atoms in these modes are shown to bend in relation to the Nitrogen atom. 4,5,6 are stretching vibrations as the bond extends and contracts bringing the Hydrogen away from and back to the central Nitrogen atom. Absorbtions that are due to the bond to bend occur at lower wavenumbers than absorbtions due to stretching as it usually requires more energy (higher frequency EM radiation) in order to make a bond stretch.
Which mode is highly symmetric?
Mode 4 is highly symmetric as all of the Hydrogens stretch by the same amount in the same direction.
One mode is known as the "umbrella" mode, which one is this?
Mode 1 is the "umbrella" mode as it involves the inversion of the molecule which is a similar to mechanism to an umbrella inverting on a windy day.
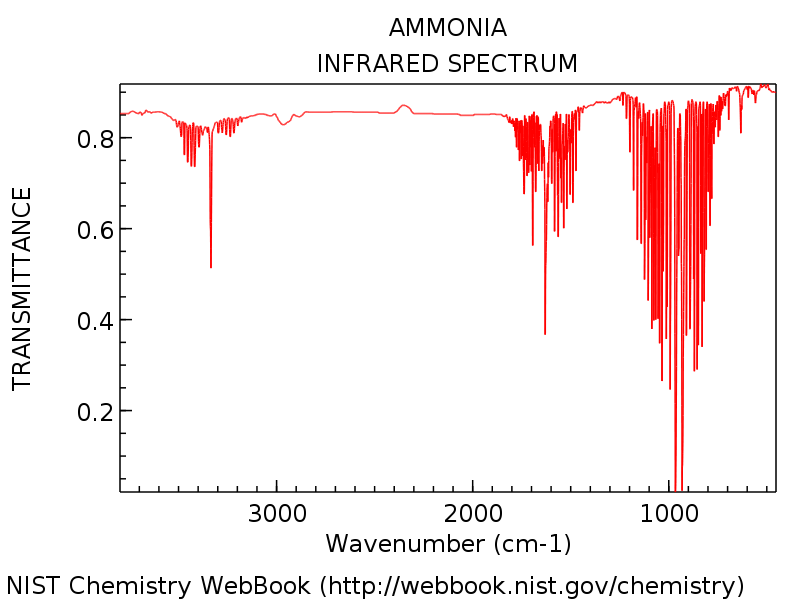
How many bands would you expect to see in an experimental spectrum of gaseous ammonia?
3 bands for (5,6), (2,3) and (1) as degenerate modes will contribute to the same band. The highly symmetric mode does not result in a change in dipole which means it will be IR inactive and therefore will not have a band associated with that particular vibrational mode. This can be confirmed as the actual IR spectrum shows three clear absorbance bands. As this is a gaseous sample of Ammonia used in the spectrometer, one can also see the bands caused by a changes in molecular rotations. The energy gap between rotational states with a different quantum number is significantly lower than the gap for vibrational modes so for each band that are many peaks for each rotational transition.
{kind=link}
N2 Molecule
Nitrogen |
Calculation Method: RB3LYP
Basis set: 6-31G(d,p)
E(RB3LYP): -109.52412868
Point Group: D∞h
N-N Bond Length: 1.10550Å (lit: 1.1Å)2
Item Value Threshold Converged?
Maximum Force 0.000001 0.000450 YES
RMS Force 0.000001 0.000300 YES
Maximum Displacement 0.000000 0.001800 YES
RMS Displacement 0.000000 0.001200 YES
Predicted change in Energy=-3.401113D-13
Optimization completed.
-- Stationary point found.
----------------------------
! Optimized Parameters !
! (Angstroms and Degrees) !
-------------------------- --------------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------------------
! R1 R(1,2) 1.1055 -DE/DX = 0.0 !
--------------------------------------------------------------------------------
{kind=link}
H2 Molecule
Hydrogen |
Calculation Method: RB3LYP
Basis set: 6-31G(d,p)
E(RB3LYP): -1.17853936
Point Group: D∞h
H-H Bond Length: 0.74279Å (Lit: 0.74Å)2
Item Value Threshold Converged?
Maximum Force 0.000000 0.000450 YES
RMS Force 0.000000 0.000300 YES
Maximum Displacement 0.000000 0.001800 YES
RMS Displacement 0.000001 0.001200 YES
Predicted change in Energy=-1.164080D-13
Optimization completed.
-- Stationary point found.
----------------------------
! Optimized Parameters !
! (Angstroms and Degrees) !
-------------------------- --------------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------------------
! R1 R(1,2) 0.7428 -DE/DX = 0.0 !
--------------------------------------------------------------------------------
{kind=link}
Reaction Energy - Haber-Bosch Process
The Haber-Bosch process was developed in the early 20th century and allowed for Ammonia, a key chemical in the manufacture of fertilizers and high explosives, to be industrially manufactured. Although the mechanism was discovered to have 6 steps, it can be summarized by a simple reversible equation for the reaction between gaseous Hydrogen and Nitrogen.
- N2 (g) + 3H2 (g) ↔ 2NH3 (g)
The change in energy for this reaction (conversion of Hydrogen and Nitrogen into Ammonia) can be calculated by subtracting the total energy of the reactants (energy that is loss/converted) from the total energy of the products. The energy of both the reactants and the products were calculated using Gaussian and then converted from atomic units to kj•mol-1.
ΔE=2E(NH3)-[E(N2)+3E(H2)]= -0.0557907 au = -146.48 kj•mol-1
The change in energy was calculated to be negative which shows the reaction is exothermic and therefore gives off heat. This implies that the products are more stable and lower in energy than the reactants as energy was released in order for Hydrogen and Nitrogen to form Ammonia.
F2 Molecule Information
Fluorine |
Calculation Method: RB3LYP
Basis set: 6-31G(d,p)
E(RB3LYP): -199.49825218
Point Group: D∞h
F-F Bond Length: 1.40281Å (Lit: 1.42Å)2
Item Value Threshold Converged?
Maximum Force 0.000128 0.000450 YES
RMS Force 0.000128 0.000300 YES
Maximum Displacement 0.000156 0.001800 YES
RMS Displacement 0.000221 0.001200 YES
Predicted change in Energy=-1.995024D-08
Optimization completed.
-- Stationary point found.
----------------------------
! Optimized Parameters !
! (Angstroms and Degrees) !
-------------------------- --------------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------------------
! R1 R(1,2) 1.4028 -DE/DX = 0.0001 !
--------------------------------------------------------------------------------
{kind=link}
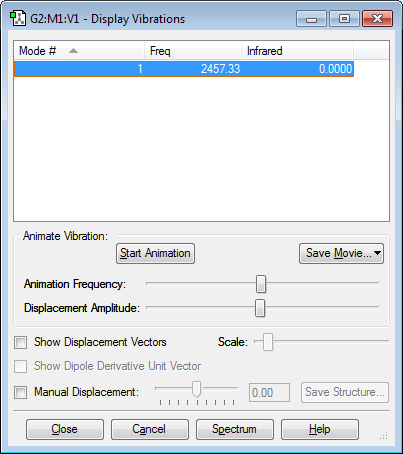
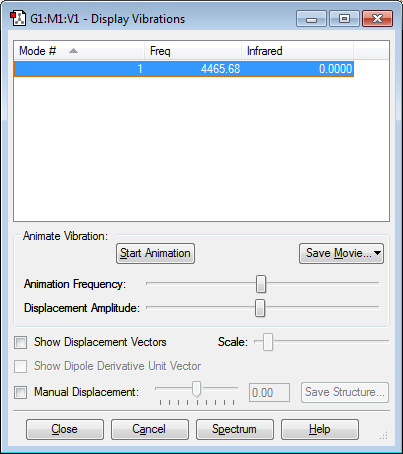
For diatomic molecules which are linear in nature, the number of vibrational modes correspond to the equation (3N-5). F2 has only one vibrational mode which is a symmetric stretch of the bond. As the molecule is a homonuclear diatomic and it's vibrational mode does not generate a dipole (transition dipole moment = 0), it is IR inactive and will not produce a peak on the spectrum. However, F2 is very reactive and unstable, which makes performing spectroscopic or other types of analysis on the molecule is very difficult.
Homonuclear diatomic molecule will not distribute charge unevenly around the molecule as a Fluorine atom is not able to draw electron density away from another Fluorine atom. Therefore the charges on the atoms in this molecule will both be 0 as there is no dipole present.
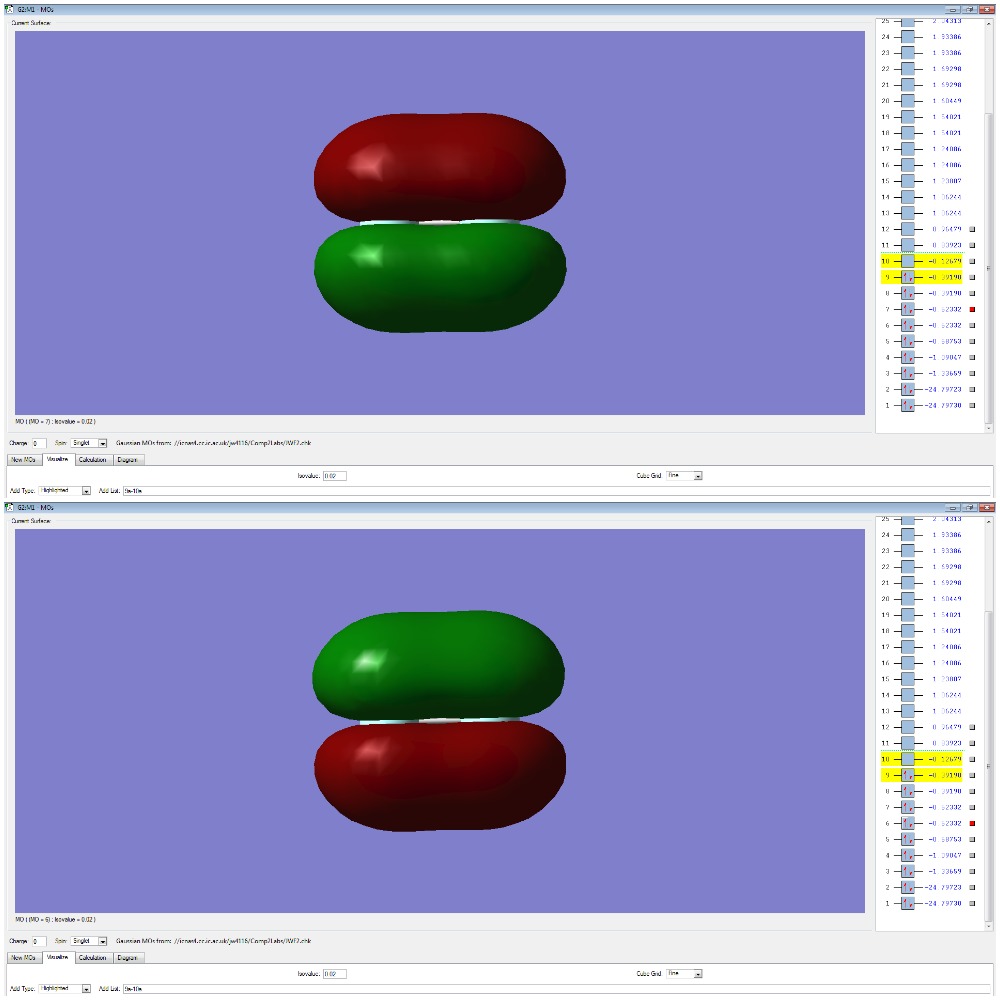
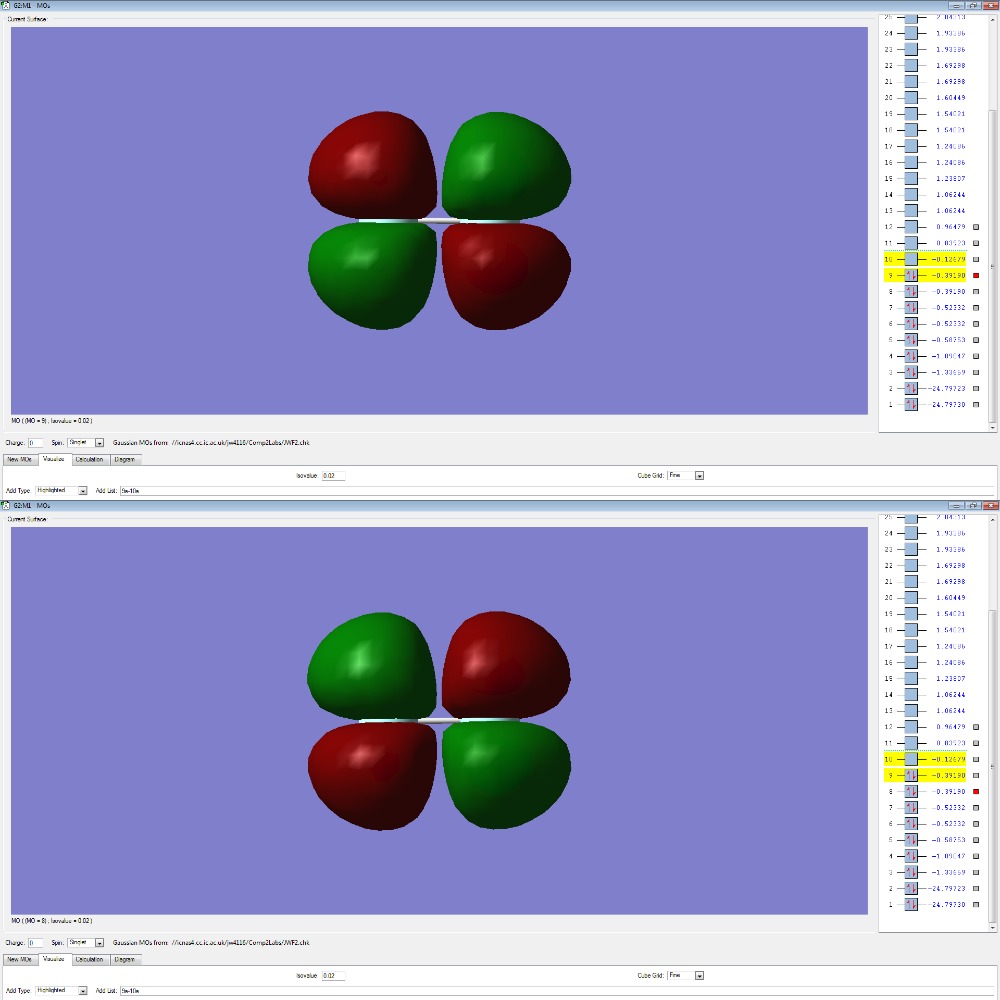
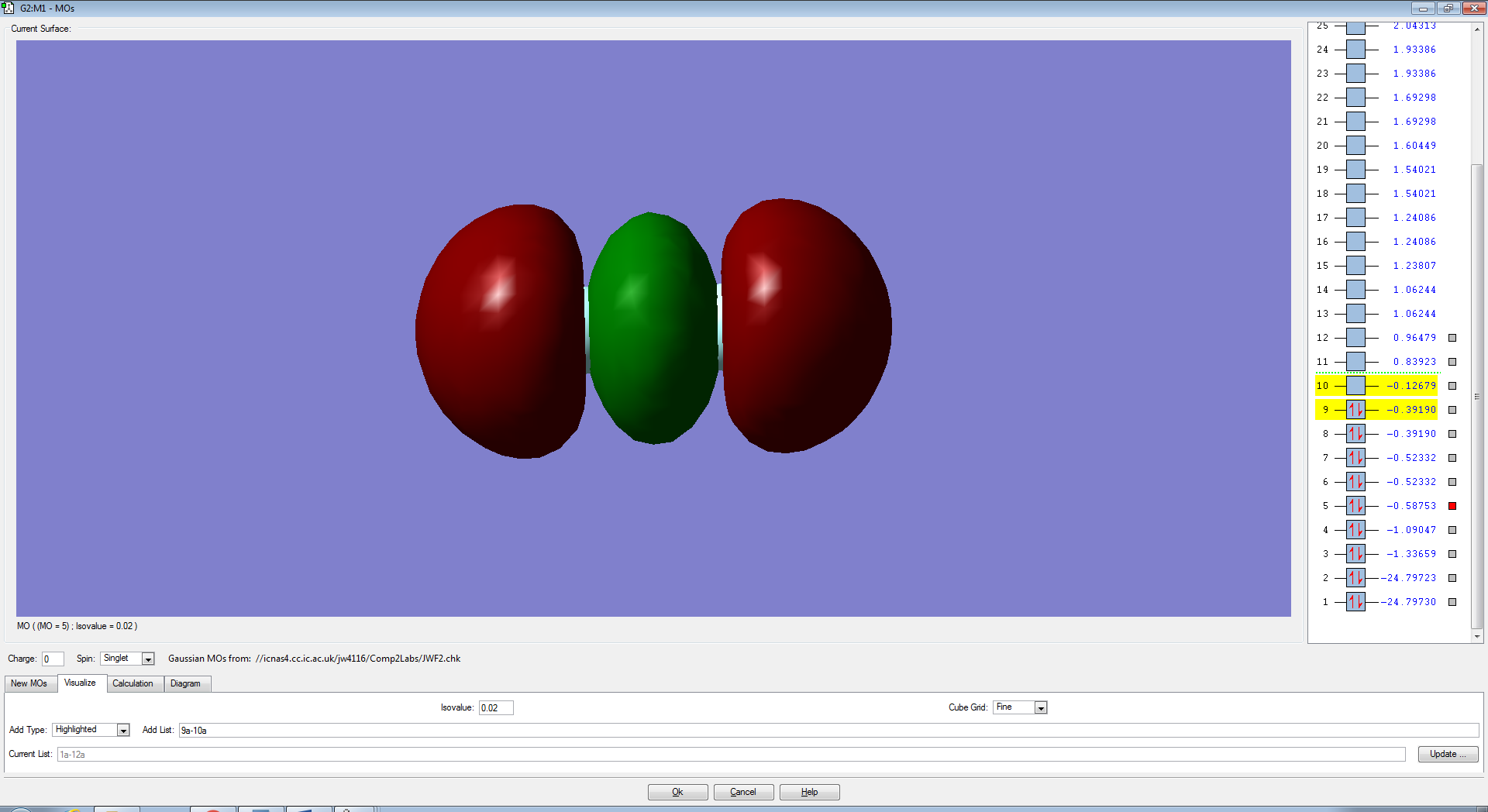
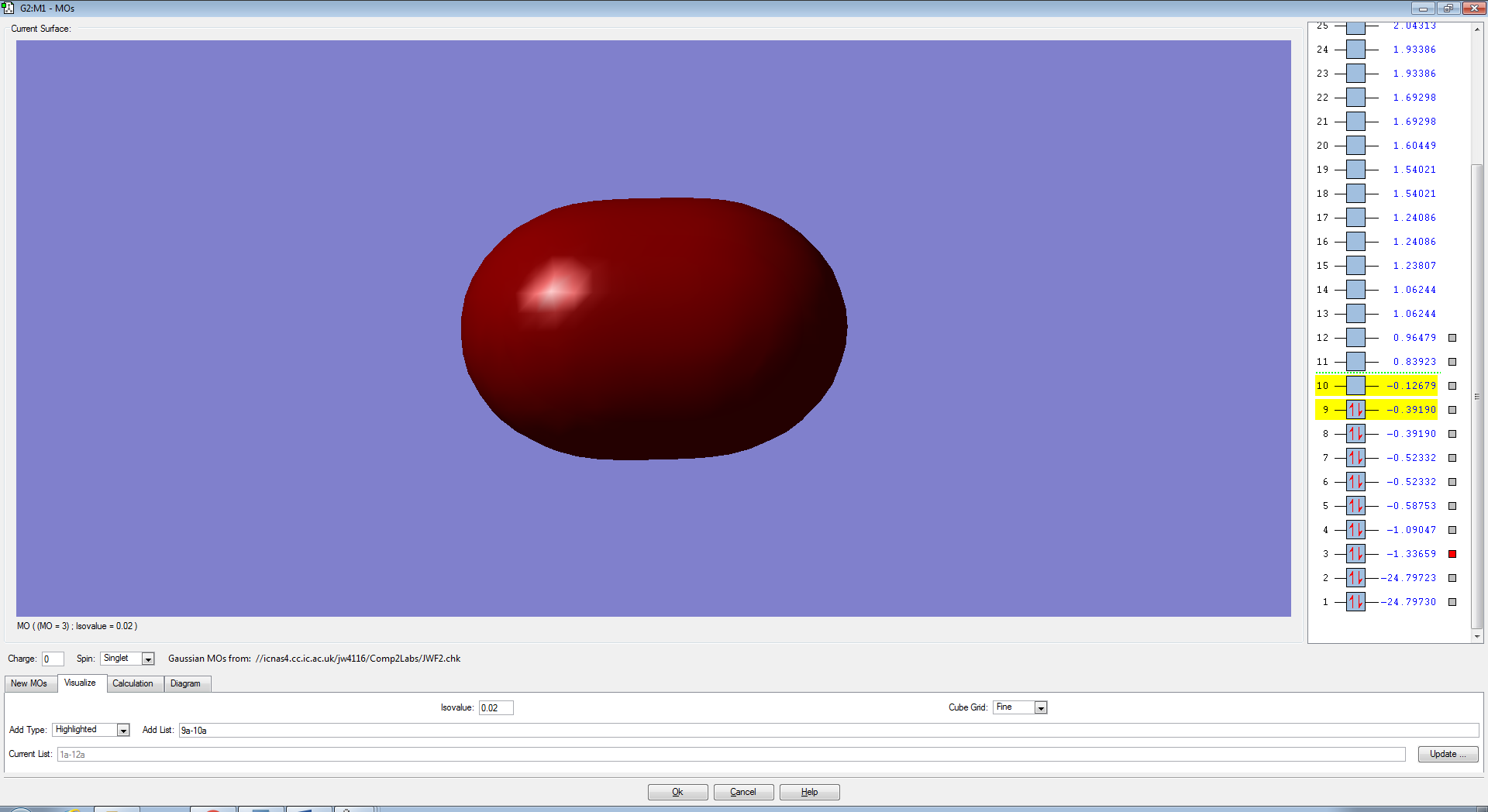
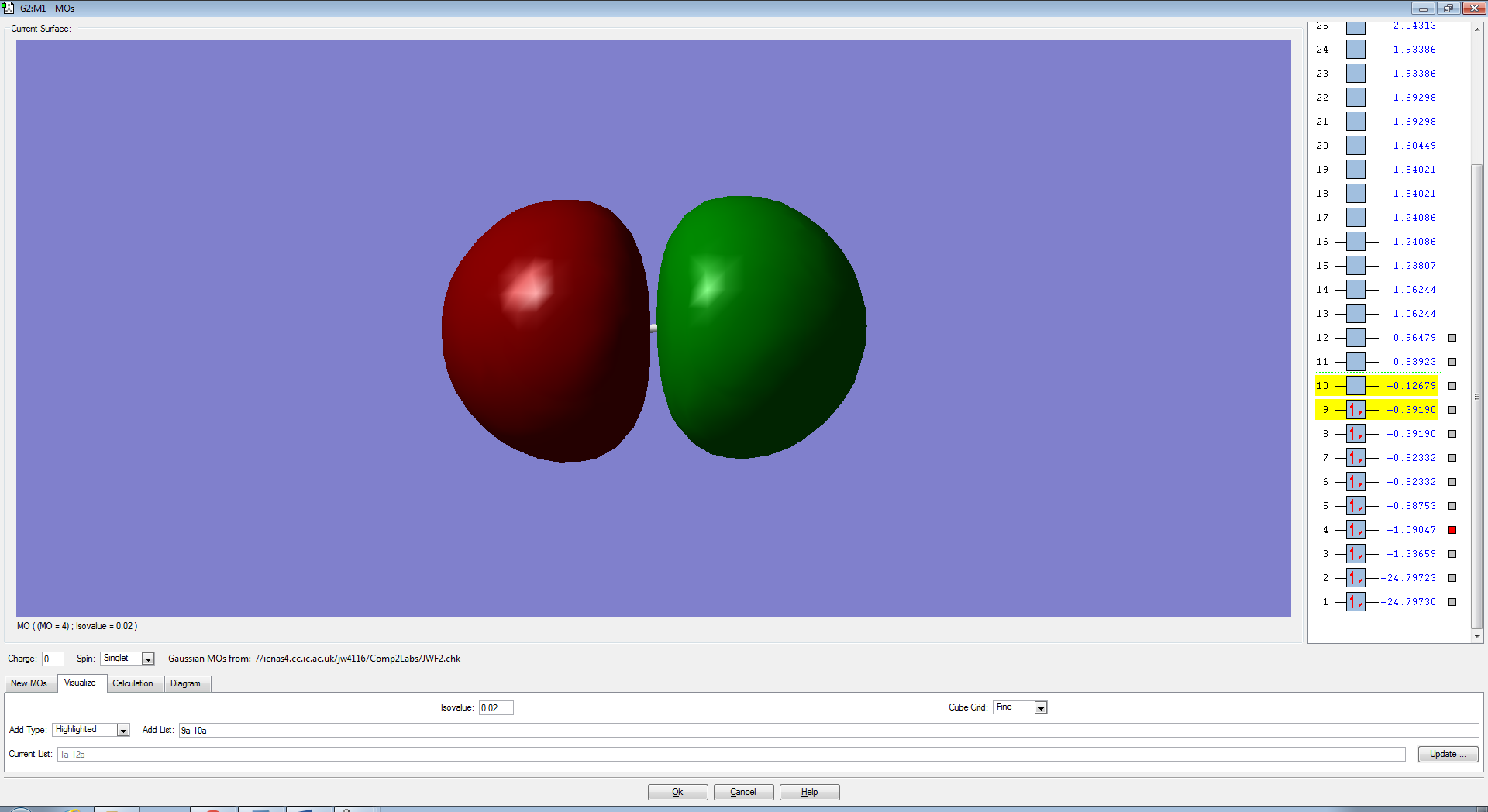
F2 Molecular Orbitals
The bond order for F2 is 1 and is defined by the equation below. This means that there is only one molecular orbital or two atomic orbitals contributing to the bond holding the atoms together.
The p-electrons present in each Fluorine atom overlap to form 4 non-bonding pi (π) orbitals and a bonding sigma (3σ).
The two degenerate π orbitals are formed from the constructive overlap of the parallel p-orbitals present in fluorine which have ungerade symmetry as the sign of the molecular orbital is changed after an inversion operation. The destructive overlap, due to the atomic orbitals being out of phase, contributes to the pi-antibonding (π*) orbitals (HOMO) which have gerade symmetry. As there are an equal number of electrons in the π and π*, the orbitals are non-bonding and will not form a bond in order to hold the two atoms together. The end to end overlap of p-orbitals result in the formation of a 3σ bonding orbital which, due to it's relative stability and lack of electrons in the 3σ* orbital (LUMO) , will contribute the most to the actual bond.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The molecular orbitals from the 3s-3s overlap bonding and anti-bonding and from subsequent molecular orbitals formed from more stable, core atomic orbitals are lower in energy than the MO's formed from the p-orbitals. Therefore the σ and σ* anti-bonding orbitals are filled first (Aufbau Principle) which results in a non-bonding molecular orbital which does not contribute to the overall bond.
{kind=link}
{kind=link}
References
1. CRC Handbook of Chemistry and Physics, 94th ed. http://www.hbcpnetbase.com. Page 9-26. (accessed March 2017).
2. Wired Chemist, http://www.wiredchemist.com/chemistry/data/bond_energies_lengths.html, (accessed March 2017).