
MRD:01357674 year2 inorganic comp lab
Year 2 Inorganic Computational Lab
EX3 section
BH3
Calculation Type : B3LYP/6-31G level
Summary Table:
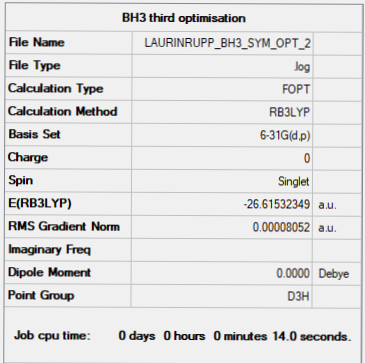
Item table:
Item Value Threshold Converged? Maximum Force 0.000161 0.000450 YES RMS Force 0.000105 0.000300 YES Maximum Displacement 0.000639 0.001800 YES RMS Displacement 0.000418 0.001200 YES
Frequency analysis .log file : Media:01357674_Laurinrupp_bh3_freq.log
Low Frequencies:
Low frequencies --- -0.2456 -0.1129 -0.0054 44.0270 45.1846 45.1853 Low frequencies --- 1163.6049 1213.5924 1213.5951
Since all the calculations have converged, and there is a significant difference between the Low frequencies and the first 'real' frequencies (45-1164 cm-1) we can conclude that the configuration we have obtained is indeed the lowest energy configuration, and not simply a local minimum in energy.
Jmole Image:
Optimised BH3 molecule |
Frequency Table:
| wavenumber (cm-1 | Intensity (arbitrary units) | symmetry | IR active? | type |
| 1164 | 92 | A2" | yes | out-of-plane bend |
| 1214 | 14 | E' | slightly | bend |
| 1214 | 14 | E' | slightly | bend |
| 2580 | 0 | A1' | no | symmetric stretch |
| 2713 | 126 | E' | yes | asymmetric stretch |
| 2713 | 126 | E' | yes | asymmetric stretch |
IR Spectrum:
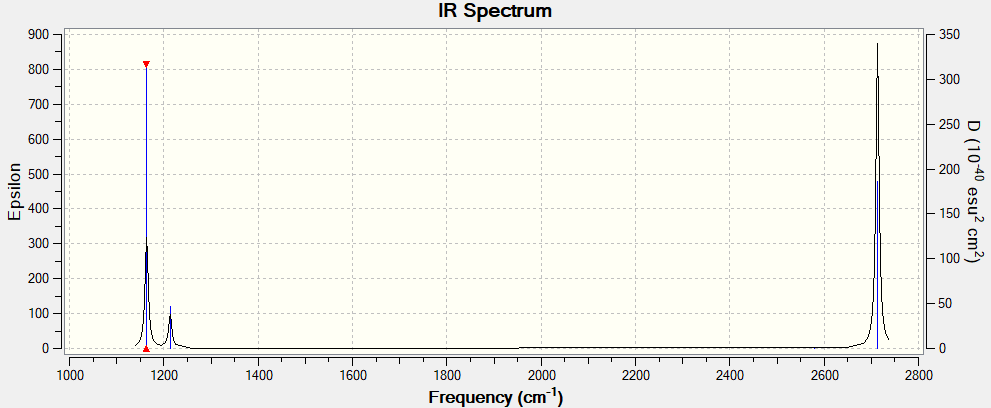
First we note that all our IR frequencies are indeed positive, suggesting that our calculations were carried out correctly. There are only 3 peaks visible on the IR spectrum as peak number 5 (at 2580 cm-1) is IR inactive due to the fact that it is a symmetric stretch that does not change the dipole moment of the molecule. Furthermore peaks 2 and 3 (at 1214 cm-1) as well as 5 and 6 (at 2713 cm-1) have almost identical frequencies, and hence overlap in the IR spectrum, leaving 3 distinct, resolved peaks.
MO Diagram:
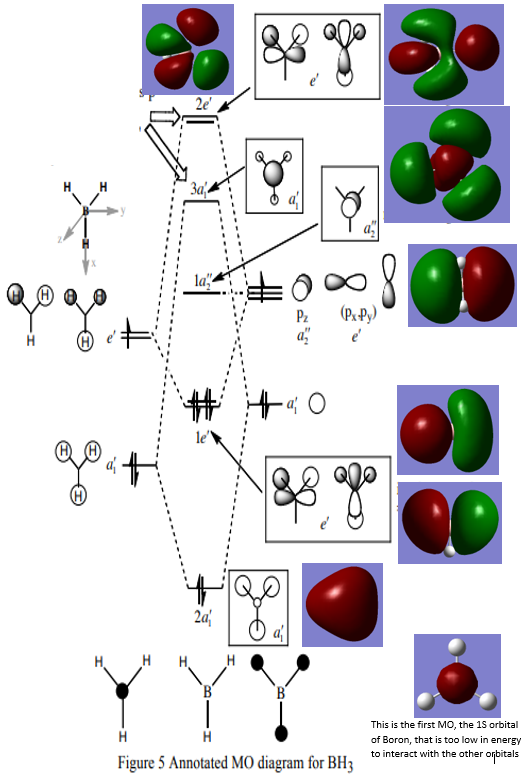
From the diagram it can be seen that up till the anti-bonding molecular orbitals the theoretical results coincide almost perfectly with the 'real' calculated results. However the 3a'1, as well as the 2e' deviate significantly. The 3a'1 orbital seems to have more p-orbital contributions than expected, and the two 2e' orbitals also have some more complex orbital contributions, probably from the p-orbitals on the boron, and maybe even the d-orbitals. This suggests that MO theory works very well for lower energy bonding orbitals, but fails to predict more complex orbital contributions for the higher energy anti-bonding orbitals.
Great discussion considering both the similarities and differences between the real and LCAO MOs. Smf115 (talk) 22:41, 16 May 2019 (BST)
NH3
Calculation Type : B3LYP/6-31G level
Summary Table:
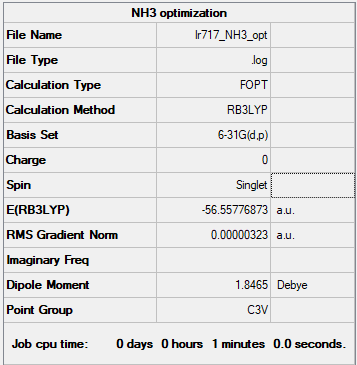
Item table:
Item Value Threshold Converged? Maximum Force 0.000006 0.000450 YES RMS Force 0.000004 0.000300 YES Maximum Displacement 0.000012 0.001800 YES RMS Displacement 0.000008 0.001200 YES
Low frequencies --- -8.5646 -8.5588 -0.0044 0.0454 0.1784 26.4183 Low frequencies --- 1089.7603 1694.1865 1694.1865
Once again, it can be seen that all the calculations have converged, and that there is a significant difference between the low frequencies and the first 'real' frequencies, confirming that this is the lowest energy configuration.
NH3BH3
Calculation Type : B3LYP/6-31G level
Summary Table:
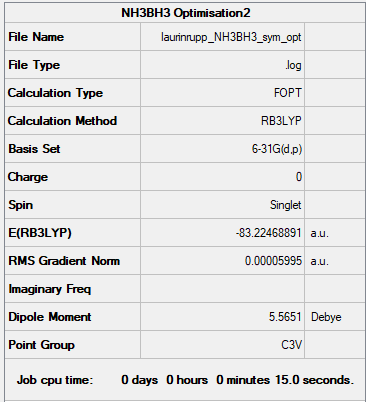
Item table:
Item Value Threshold Converged? Maximum Force 0.000122 0.000450 YES RMS Force 0.000058 0.000300 YES Maximum Displacement 0.000531 0.001800 YES RMS Displacement 0.000296 0.001200 YES
Low frequencies --- -0.0573 -0.0499 -0.0073 21.6972 21.7073 40.5564 Low frequencies --- 266.0216 632.3607 640.1371
As before, a minimum energy configuration has been found.
Ok structure information and good remark about the low frequencies in the BH3 section. However, the frequency log files and jmols are missing for NH3 and NH3BH3, and the file submitted for BH3 is actually your input file. Smf115 (talk) 22:44, 16 May 2019 (BST)
Calculations
From the molecular analysis carried out above we can calculate the dative bond strength of B-n in NH3BH3. First of all we report the energy of the fragment in a.u.:
E(NH3)= -56.55777 a.u.
E(BH3)= -26.61532 a.u.
E(NH3BH3)= -83.22469 a.u.
Hence the association energy is equal to:
ΔE = E(NH3BH3)-[E(NH3)+E(BH3)] = -0.05160 a.u.
Now to convert from a.u. (hartree) to kJ/mol we simply multiply by 2625.5 kJ/mol, so: ΔE = -135 kJ/mol
This is a reasonably weak bond, as a 'normal' B-N bond dissociation energy is around 389 kJ/mol (note that since we calculated a bond association energy the signs are reversed). This is probably due to the fact that BH3 is already quite a stable molecule, and since it is so small, adding a bond leads to more electron-electron repulsion. Hence the dative bond results in less stabilisation of the molecule, making it a weak bond.
Good presentation of the energies, correct calculation and nice consideration of the accuracy of the final reported value. The comparison to the B-N bond is good but you should have a reference for any literature value. The idea raised in the discussion is interesting but not quite right. Smf115 (talk) 22:50, 16 May 2019 (BST)
NI3
Calculation Type : B3LYP/Gen
Summary Table:
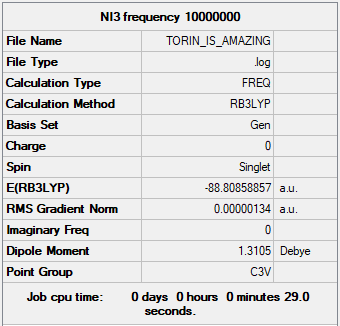
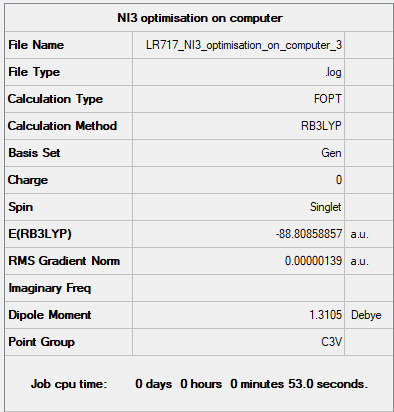
Item table:
Item Value Threshold Converged? Maximum Force 0.000002 0.000450 YES RMS Force 0.000002 0.000300 YES Maximum Displacement 0.000022 0.001800 YES RMS Displacement 0.000014 0.001200 YES
Optimisation .log file : Media:LR717_NI3_OPTIMISATION_ON_COMPUTER_3.LOG
Frequency analysis .log file : Media:01357674_TORIN_IS_AMAZING.LOG
Low frequencies --- -12.5522 -12.5460 -6.0047 -0.0039 0.0191 0.0664 Low frequencies --- 100.9969 100.9977 147.3377
It can be seen that all calculations converged, and that there is a less pronounced difference in low frequencies, probably due to the heavy Iodines vibrating at lower frequencies.
Jmole Image:
Optimised BH3 molecule |
From the analysis it was found that the average N-I bond distance is equal to 2.1840 Å, which is quite a long bond as expected as the Iodine is in period 5, and has large diffuse electron shells that lead to stonier electron-electron repulsion with close, neighbouring molecules, so that the equilibrium bond length is increased (also suggesting a weaker bond strength).
Project Section
N(CH3)4+
Calculation Type : B3LYP/6-31G level
Summary Table:
Item table:
Item Value Threshold Converged? Maximum Force 0.000011 0.000450 YES RMS Force 0.000004 0.000300 YES Maximum Displacement 0.000054 0.001800 YES RMS Displacement 0.000023 0.001200 YES
Frequency analysis .log file : Media:01357674_N(CH3)4_FREQ.LOG
Low Frequencies:
Low frequencies --- 0.0006 0.0006 0.0009 34.4418 34.4418 34.4418 Low frequencies --- 216.7307 316.0807 316.0807
It is shown that the calculations for our molecule converged, and that there is a significant difference in low frequencies. The 'real' frequencies start quite low because the CH3 groups are reasonably large and heavy, leading to lower frequency bond vibrations.
List of IR Frequencies:
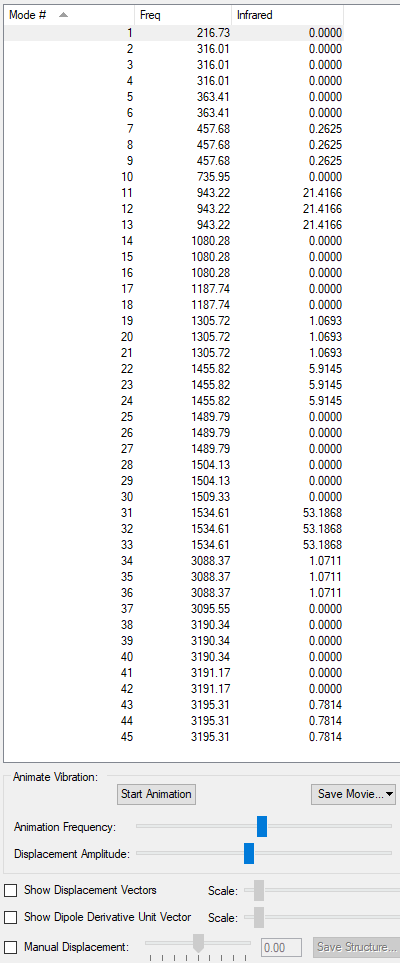
This shows that there are no negative IR frequencies, reinforcing that there were no errors in our calculation.
Your log file for your frequency calculation on [N(CH3)4]+ is correct and has the correct charge. However, in the information, you've included the optimisation summary table which also shows the wrong charge (0) for the molecule. The long list of frequencies isn't necessary to include, although good knowledge showing that you don't want your structure to have an imaginary frequency (as then not at a stable minimum). Instead of listing all of the frequencies, this information is actually displayed in the summary table for the frequency calculation (see your one below) which is why we wanted the frequency summaries. Smf115 (talk) 22:04, 19 May 2019 (BST)
P(CH3)4+
Calculation Type : B3LYP/6-31G level
Summary Table:
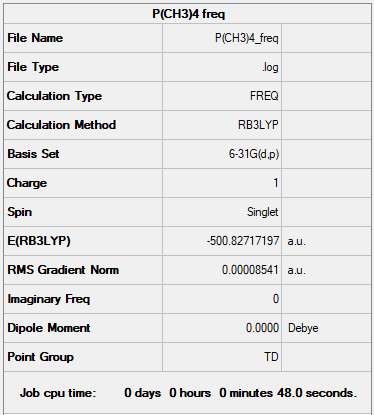
Item table:
Item Value Threshold Converged? Maximum Force 0.000143 0.000450 YES RMS Force 0.000033 0.000300 YES Maximum Displacement 0.000622 0.001800 YES RMS Displacement 0.000271 0.001200 YES
Frequency analysis .log file : Media:lr717_P(CH3)4_FREQ.LOG
Low Frequencies:
Low frequencies --- -0.0015 -0.0015 0.0011 51.3641 51.3641 51.3641 Low frequencies --- 186.6817 211.5017 211.5017
As for N(CH3)4+, the calculations converge, and the difference in frequencies is slightly less as expected, since phosphorus is a heavier atom, decreasing the bond stretching frequencies.
List of IR Frequencies:
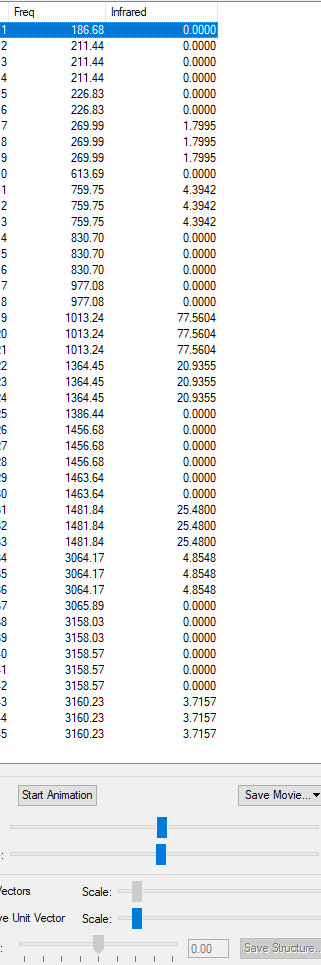
It is shown that there are no negative IR frequencies, which would have meant a mistake was made in the calculations.
Results and Discussion
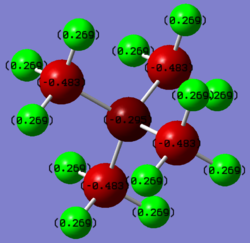
Using gaussview, a charge analysis of both the molecules was done, and the result can be seen below.
NB: Gaussian is the program which does the calculations and GaussView is just a GUI to visualise the structures and results. Smf115 (talk) 08:16, 20 May 2019 (BST)
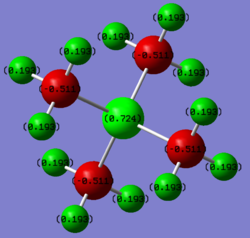
The smallest possible charge range was chosen to visualise both molecules (-0.511 to 0.724). This was done to make the difference in charges as apparent as possible, in order to be able to visualise the different charge distributions better. It can be seen that for N(CH3)4+, the central Nitrogen atom actually has a negative charge. This is because Nitrogen is quite electronegative, as it has a high effective nuclear charge (because it is a second row element with 7 protons), resulting in very contracted electronic orbitals, and a tendency to pull electrons towards it. The reason it is only slightly negative in this case is due to the positive charge negating most of these effects, and effectively taking electron density away from the Nitrogen. Interestingly, the Carbons have the most negative charge because they are more electronegative than the Hydrogens, and thus can pull electron density from all 3 of them (whilst at the same time the Nitrogen is pulling electron density from the Carbons). This leaves the Hydrogen atoms being quite electropositive, and having the positive charge distributed amongst them.
In contrast, the Phosphorus in P(CH3)4+ is very electropositive. That is because it is similarly electronegative to the Hydrogens, as it is a third row element, having much larger and more diffuse orbitals (furthermore as it doesn't have any d-orbitals it doesn't experience d-orbital contractions). This means that the Phosphorus is quite polarisable, and carries a lot of the positive charge. In contrast the Carbons are even more electronegative as they can now pull electron density from both the Hydrogens and the Phosphorus, thereby also making the Hydrogens less electropositive.
Nice consideration of the nature of the P orbitals compared to H, however, you've used the Mulliken charges for the charges and not the NBO ones resulting in the wrong values/distribution. The electronegativity argument is good and you discuss all the atoms, but other effects such as symmetry could have been considered and in some places, your arguments aren't quite correct/clear (e.g. what do you mean exactly that the positive charge negates the effects?). Smf115 (talk) 08:16, 20 May 2019 (BST)
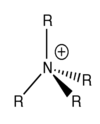
In the traditional Nitrogen cation representation (as seen on the right), the positive charge is often shown to be on the Nitrogen itself. This is because if one calculates the formal charges for the atoms in the molecule. Nitrogen has a formal charge of +1. Essentially this model is saying that we simply remove one valence electron from the Nitrogen (leaving it with an electronic configuration of 1s2,2s2,2p4) so that it can from 4 bonds, but only have 8 valence electrons. However, we have shown that this is not the case, and that in fact the positive charge is located on the hydrogens of the R group for this molecule.
An ok comparison between the traditional picture and the real charges, however, to improve you could have been clearer about how the formal charge is actually calculated. Smf115 (talk) 08:16, 20 May 2019 (BST)
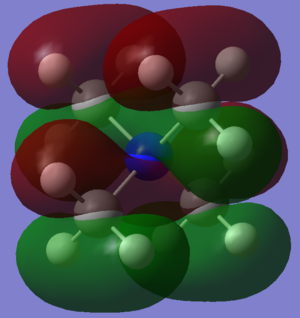
Finally, three Molecular Orbitals (MOs) of the N(CH3)4+ molecule were chosen, and their potential Linear Combination of Atomic Orbitals (LCAO) was worked out. The results are illustrated below:
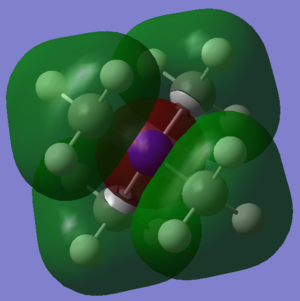



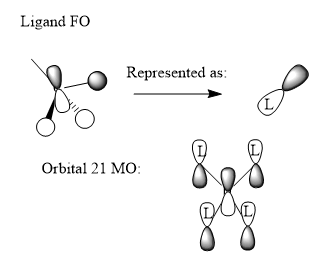
First of all, MO number 10 has been pictured. It is clearly seen that it has 4 symmetrical triangular shaped orbitals on the outside where the CH3 groups are, and a large, almost spherical centre of opposite phase around the central Nitrogen. Looking more closely at the MO, it can bee seen that there is a phase boundary node on each of the carbons. This suggests that a Carbon P-orbital is involved. As a result, the LCAO diagram on the right was produced. It states that the Carbon P orbital pointing directly towards the Nitrogen combine with the Hydrogen 2s orbitals, and then bond all together to the central Nitrogen 2s orbital. In reality, we would also expect some contribution from the Carbon 2s orbital, as well as contributions from the Nitrogen 3s. The interactions in the CH3 groups are definitely bonding, as well as the interactions between half of the Carbon p-orbital that is in phase with the central Nitrogen s-orbital, making this a bonding orbital overall. However, spatially the Hydrogens of opposite phase are still reasonably close to the central Nitrogen s-orbital of opposite phase, resulting in some weak anti-bonding interaction between the green and red phases.
The second MO that can be seen is number 15, whose orbitals have a centre of inversion on the Nitrogen. Looking back at our MO diagram of BH3, each ligand resembles somewhat the first 1e' molecular orbital. That is to say the in phase overlap of a Carbon p-orbital with two Hydrogen s-orbitals of opposite phase, whilst the third Hydrogen does not contribute (because that would change the symmetry). In this case, another orthogonal p-orbital that points towards the non-bonding Hydrogen atom interacts with the previous combination, resulting in two elongated lobes that are slightly asymmetric, with a larger orbital on one side compared to the other. The 4 methyl groups then interact in the full molecule MO, but there is no contribution from the Nitrogen. Each methyl group overlaps with two other methyl groups in that are in phase, creating the MO that is seen. The hydrogens in each CH3 groups have opposite phase, but are also quite far apart so that they are only slightly anti-bonding. The Carbon p-orbitals mostly overlap positively with the Hydrogen s-orbitals, with only a little repulsion at the phase boundary. The methyl groups also have positive overlap between each other, however there is quite a bit of anti-bonding character through space as the lobes of opposite phase are in very close proximity to each other in all directions. Overall one would think the direct bonding interactions outweigh the spatial anti-bonding interactions, however this is not certain, and this orbital could be non-bonding.
Lastly, one can see MO number 21, the HOMO of this molecule. Although it looks very symmetric, it actually only has C2 symmetry as well as a σv symmetry plane. The methyl groups remind us of the BH3 MO again, this time a distorted version of the second 1e' orbital. It is an in phase combination of two 1s Hydrogen orbitals with one lobe of a Carbon p-orbital, and the remaining 1s Hydrogen orbital of opposite phase with the other lobe. However, due to the distorted geometry (it is no longer planar) this overlap is not as favourable. Each of the methyl groups then overlaps in phase with a central Nitrogen 2p-orbital creating the observed orbital. As for the other orbitals, although the direct overlap is favourable between the Carbons and the Hydrogens, as well as the methyl groups and the Nitrogen, there is significant anti-bonding character as spatially these lobes of opposite phase are very closely packed, especially in the centre around the Nitrogen, causing electron-electron repulsion and destabilising the molecule. Overall this orbital is probably still bonding, but only weakly.
Good range of MOs chosen and you've obviously really considered the FOs and the interactions present in the LCAOs. However, although you have correctly tried to apply the BH3 MO diagram to CH3, your FOs aren't correct for MO 10 and 15. For example, MO 15 is just the e' FO which you identified and there is not second p orbital on the C. To improve, the analysis and description is good but presenting the interactions through annotations on the diagrams (such as in the Inorganic course notes may make it easier for you to analyse and for someone else to understand. Smf115 (talk) 21:34, 21 May 2019 (BST)
Overall an ok report which was let down by missing/incorrect structure files in section 1. Smf115 (talk) 21:34, 21 May 2019 (BST)