
Jn307Module1:h1iamachemist
The Hydrogenation of Cyclopentadiene Dimer
Endo and Exo Forms


The dimerisation of Cyclopentadiene occurs by the incoming cyclopentadiene attacking either pointing towards or pointing away from the other Cyclopentadiene ring which would results in an endo formation if pointing in the same direction or anti to each other which would result in an exo formation. This is illustrated in the diagram below. In the Molecular mechanics used, the energy of the endo formation is higher which suggests a greater steric strain due to the two cyclopentadiene pointing in the same direction as each other. However, in the exo form, the two cyclopentadiene are pointing away from each other which allow the molecule to have more steric freedom and hence should be lower in energy.
The molecular mechanics used in chembio3D indicates that the exo dimer of cyclopentadiene have a total energy of 32.883 kcal/mol while the endo dimer of the cyclopentadiene has a total energy of 34.0061. This supports the Alder-Endo rule that the exo product is the most stable form with the lowest energy; although the endo product is the major product. As the picture on the right shows, the endo dimer atoms are much more situated closer to each other than the exo form which will give the endo dimer more steric hindrance and therefore would be higher than energy than the endo form. The exo form is therefore the most thermodynamic stable product with the lowest energy, however the endo dimer is the kinetic product which means that it has the lowest activation energy than the exo dimer. A lower activation energy would means that this product would form first in a reversible reaction however if the reaction if reversible, with enough time and energy, the reaction can go back and form the more thermodynamic product. The dimerisation of Cyclopentadiene is therefore kinetic controlled rather than under thermodynamic control.
Hydrogenation of the Endo Cyclopentadiene dimer


| The Relative contributions of the Dihydro Derivatives | ||
| Relative Contributions | Dihydro Product 3/kcal/mol | Dihydro Product 4/kcal/mol |
|---|---|---|
| Stretch | 1.2794 | 1.1245 |
| Bend | 19.0909 | 13.0210 |
| Stretch-bend | -0.8432 | -0.5648 |
| Torsion | 11.1365 | 12.4238 |
| Non-1,4 VDW | -1.6454 | -1.3323 |
| 1,4 VDW | 5.7864 | 4.4420 |
| dipole/dipole | 0.1622 | 0.1409 |
| Total Energy | 34.9668 | 29.2551 |
Product 4 has the lowest energy out of the two with 29.2551 kcal/mol compared with the energy of product 3 of 34.9668 kcal/mol. In a thermodynamic sense, product 4 would be the most stable as it has the lowest energy, and if the activation energy is the same, product 4 would form as the major product. The stretching of both product 4 and product 3 are very similar with product 3 slightly higher, this suggests that product 3 would need more energy to stretch which may be because the double bond is closer to the bridging carbon and so there is not enough space for the bond lengths to stretch before the steric hinderance overrides the stretching energy. The torsion energy of product 4 is 1.2873 kcal/mol bigger than product 3 which may suggests that the double bond in product 4 prevents it from being allowed to twists in a particular conformation and therefore has a more rigid conformation. The VDW contribution of product 3 is higher than product 4 suggesting that product 3 is slightly more polar than product 4 and therefore is more polarised which may because the bridging and the double alkene are on the same side and hence increases the electron density on that side while in product 4, the electrons are spread out and hence the van der Waals forces are a much smaller value than product 3.
Stereochemistry of Nucleophilic additions to a pyridinium ring (NAD+ analogue)
The Alkylation of the Pyridine Ring with Methyl Magnesium Iodide


By trial and error and minimising the energy of the molecule 5, the lowest total energy was found to be 43.0536 Kcal/mo and the carbonyl in a slightly up position as it can be seen in the jmol file. The carbonyl is very close to the equatorial position where it is not hindered by any atoms. The dehedral angle it also prefers to be is at -171°, which is around 60° if only three atoms were considered. This is the optimum angle for a sp2 triangular planer configuration. The below table shows that by moving the carbonyl oxygen in various positions, the carbonyl would usually returns to the optimum position where the angle is maximased and the energy is minimised. However, in certain situations, the carbonyl would adopt an axial position. In the jmol button shows, this oxygen is now very close to the hydrogen on the other side of the ring, and would experience a higher steric hinderance and therefore a higher energy which was found to be around 160 kcal/mol. This is almost 4 times the energy of the minimum energy and suggests that due to the high energy, this product with a carbonyl oxygen in a axial position would not form.

| Angle of the carbonyl in relation to the ring | dihedral angle after minimising | Total Energy/kcal/mol |
|---|---|---|
| -171 | -171 | 43.0536 |
| 52 | 175 | 43.0789 |
| 146 | -117 | 43.0761 |
| 21 | 59 | 161.2013 |
| 42 | 64 | 168.1625 |
The Mechanism for the Grignard reaction

Looking at the jmol of compound 5, the carbonyl oxygen can be seen pointing slightly up. The Grignard magnesium is very electropositive while the methyl is electron rich. This would lead to a fast reaction with the pyridine ring. The oxygen is more electronegative than the methyl and therefore the magnesium would bind with the oxygen as the methyl attacks the 4-position. The methyl cannot attack the carbons because when the magnesium binds with the oxygen, the methyl would be locked and cannot move any closer to any other atom. This methyl would therefore be delivered in an up position and so both the carbonyl and the methyl would be pointing up. The iodide ion would attack the magnesium and the oxygen would reform to make the carbonyl group again.
Nucleophilic additions to a pyridinium ring

Molecule 7 is similar to molecule 5, the carbonyl prefers to be in an equatorial position which can be seen the jmol above. After much trial and error, a dihedral angle of 154° with a total energy of 63.58 kcal/mol was found. This is similar to molecule 5, and shows that there is no increased steric hinderance compared with molecule 5. Moving the carbonyl in several positions and minimising the energy would again almost lead back to the minimum energy found however if the carbonyl was moved in such a way in which the bond cannot move, the carbonyl would instead go to an axial position. In this position, the entire molecule will twists and the jmol diagram shows that the oxygen is now directly next to the bulky methyl rings as well the benzene rings, which increased the total energy to 191.74 kcal/mol; a huge jump in energy. The carbonyl clearly does not like being in the axial position and hence, this compound will not form with a carbonyl in the axial position.
-
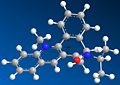 molecule 7 at highest minimisation
molecule 7 at highest minimisation -
 molecule 7 with carobonyl at axial position
molecule 7 with carobonyl at axial position


The Mechanism for the transfer of the NHphenyl to the pyridinium ring

In this example, the carbonyl oxygen is slightly pointing down as it can be seen in the jmol diagram above however, in this case the NHPhenyl does not have an electropositive group and so the mechanism to form product 8 would not occur in the same manner as the Grignard reaction. Instead, this reaction is affected by the sterics the NHPhenyl might feel if it attacks from above or below the ring. In this example, the carbonyl would act as a repulse molecule, pushing the NHPhenyl away and preventing the molecule from attacking from above. This would only leave one way that the NHPhenyl can attack which is the top face and away from the carbonyl oxygen. Therefore the NHPhenyl would form in a top face rather than the bottom face of the molecule to avoid any steric clashes that it might have felt.
In each case, the simple models might be able to be improved by adding a functional group on the carbonyl, this would make the side on the carbonyl much more bulker and it would help to investigate whether the grignard reaction will occur on the top face again and whether it can prevent the NHPhenyl from attacking should the substituent on the carbonyl be big enough.
Stereochemistry and Reactivity of an Intermediate in the Synthesis of Taxol.
-
 Molecule 9 - E= 48.9081 kcal/mol
Molecule 9 - E= 48.9081 kcal/mol -
 Molecule 10 - E = 44.2999 kcal/mol
Molecule 10 - E = 44.2999 kcal/mol


In drawing intermediates 9 and 10 in chembio3D, it was important to place the cyclohexane into a chair conformation in order to minimise the energy to the lowest total energy. it was also necessary to check that the conformation drawn is the same as the script and that the lowest possible energy can be found in the molecule after minimising. Preliminary analysis of the two intermediates would suggests that molecule 10 would be the one lowest in energy as it is much closer to the cyclohexane which is pointing away from the carbonyl which would minimise the steric clash it might feel. In molecule nine though, the carbonyl is pointing up and is directly next to the bridging methyl which would clash with the carbonyl more than the cyclohexane which would result in a higher energy and therefore higher instability. However, as both are intermediates of the synthesis of taxol, the energies of both molecules should not be that far apart.
Calculation done using MM2 suggests that molecule 10 with the carbonyl pointing down is lower in energy at 44.2999 kcal/mol than molecule 9 with an energy of 48.9081 kcal/mol which agrees with the initial assessments of the molecules. The bridging carbons would have lead to a much higher transannular interaction with the carbons than the cyclohexane which would lead to a higher energy level.
In looking at the alkene groups and following literature about bridgehead olefins, the dihedral angle of molecule nine is 156° while molecule 10 has an angle of 152° which is a significant difference in a normal dihedral angle of 180° planer molecule. This would suggests that the R groups around the double bond is slightly distorted and display an sp3 type of orbital rather than a conventional sp2 shape. In Maier and Schleyer[2] work on the ‘Evaluation of the stability of bridgehead Olefins’ they showed that the frontier molecules is slightly faster away from each other due to the steric clash that would be experience by the carbon attached to the bridgehead. This systematic diagram from the journal can be seen below.

In our system, and with the data analysed, the angle would suggests that in the 10 membered ring, the frontier molecules are not quite sp2 nor is it totally sp3[3] orbitals but rather something in between of these two. However, the angles do not have as perfect as an overlap and so it has suggested that it would raise the HUMO and lowers the LUMO and allowed electrophilic attack due to the increased twist of the double bond. however , this journal only examines small to median rings of which transannular interaction would play a large part in the instability of the bridgehead olefins. [4]
In 8, 10 and even larger rings member rings[5], it is seen from previous molecular models that the bridgehead olefins at higher rings have less strain than the same molecules with no double bond and hence is more stable. The data can be calculated using molecular mechanics and finding out the olefin strain energies if this experiment was taken further. The olefin strain energies can be calculated by the difference of the olefin strain and the saturated hydrocarbon equivalent. This would occur because of the much greater space that the molecule can afford by being much further away from the substituents and hence there is a reduced transannular effect and that the double bond is in a sp2 hybridised form rather that a saturated sp3 orbital which would involve another hydrogen and hence the transannular interaction would be less in the olefin rather than the saturated ring. This would result in a slower functionalisation of the alkene.
Modelling Using Semi-empirical Molecular Orbital Theory.

The Molecular obitals of Molecule 12
-
 HOMO
HOMO -
 LUMO
LUMO -
 LUMO+1
LUMO+1 -
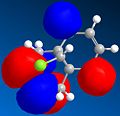 LUMO+2
LUMO+2




The molecular orbitals shown above, shows that the alkene bonds endo (closest) to the chlorine atom has a more electron density rich HUMO than the other double bond and hence would be more necleophilic and more willing to attack an electrophile. The MOPAC therefore does discriminate between the two alkene bonds. Ciepaks[6]theorised that the sigma of the C-Cl bond would bond with the σ* of the C-Cl hence lowering the energy of the alkenes, however, the sigma bond can align with both alkene bonds and does not explain why the molecule endo to the chlorine is a better nucleophile than the double bond exo to the molecule. Brian Halton[7] proposes that as well as the orbital overlap having an effect, Electrostatic π-facial asymmetry may also be used to explain the large difference in nucleophility of the double bonds.The journal suggests that the C-Cl σ* orbital is close enough with the exo C=C π orbital to overlap and hence making the C=C more stable. Brian shows that the exo C=C bond length to the bridge carbon appears to shorten to 0.24 anstroms smaller than the endo distance which is due to the increased bond order of the alkene. The endo alkene would therefore be higher in energy compared with the exo alkene and as a frontier orbital will donate it’s electrons more readily.
Part 2: Vibrational Frequency of Molecule 12 and the hydrogenated molecule 12
| Molecule | stretch/cm-1 | |
|---|---|---|
| C-Cl | C=C | |
| Molecule 12 | 548.83 | 1737.12 |
| 770.91 | 1757.37 | |
| Hydogonated Molecule 12 | 426.37 | 1753.76 |
| 528.66 | ||
As the table above shows, the stretch at 1737.12 cm-1 indicates the C=C exo to the Chlorine atom, this stretch is removed when the exo double bond is hydrogenated. When the exo double bond is hydrogenated, the endo double bond goes from 1757.37cm-1 to 1753.76cm-1, a decrease of 3.61cm-1. The energy is directly inversely proportional to the wavelength frequency therefore, when the exo double bond was hydrogenated, the endo double bond slightly stabilised. This may have been because initially, the π C-Cl would have bonded with the σ* bond, which made the bridging carbon to the exo double bond smaller. In turn, this may have made the chlorine closer to the endo double bond and thus there would have been more σ donation to the σ* C=C endo bond and hence making it slightly weaker. When the exo double bond was hydrogenated, the π->σ* system would have been lost and so the distance of the bridging carbon would return to normal and the σ->σ* bond would have weaken which in turn would have made the C=C bond stronger. In terms of the C-Cl bond, again the bond becomes stronger after the double bond was hydrogenated. This may have been because the chlorine was able to vibrate faster and stronger because it did not have to do any π donation and have lost half of the σ donation to the exo double bond.
Mini Project - Palladium-Catalyzed Intramolecular 5-exo-dig Hydroarylations of N-Arylpropiolamides[8]

As the molecule is a geometric isomer to each other, the only way to distinguish each other is a method that can differentiate between the positions of the groups on the double bond. Therefore a mass spectrum would be useless as will a optical rotation because they both do not distinguish between the two different E and Z forms. A Gas chromatography would also not help because while it may help in position isomers, it will not help to distinguish between the different positions across a double bond. A proton NMR can be used to distinguish between the two different isomers as mentioned in the journal by Hassner[9] and Davis[10] who claims that the proton on the olefin will have a slightly but noticeable change around the double bond of around 4Hz between the proton across the double bond, the trans proton would be more down fielded than the cis proton.
13-Carbon NMR of the Minor Cis ProductDOI:10042/to-2961

| GIAC NMR/ppm | experimental NMR data/ppm | Molecule number |
|---|---|---|
| 25.84 | 25.9 | 12 |
| 105.22 | 107.8 | 1 |
| 115.58 | 118.9 | 4 |
| 117.78 | 121.8 | 3 |
| 121.33 | 124.3 | 5 |
| 123.73 | 126.3 | 7 |
| 124.49 | 128.2 | 15 |
| 125.42 | 128.8 | 2 |
| 125.43 | 130.4 | 17 |
| 128.46 | 131.9 | 16 |
| 130.54 | 133.8 | 18 |
| 130.75 | 137.0 | 13 |
| 133.00 | 142.3 | 14 |
| 138.16 |
166.1 |
6 |
| 138.27 | 11 | |
| 160.32 | 8 |
13-Carbon NMR of the Major Trans Product DOI:10042/to-2962

| GIAC NMR/ppm | experimental NMR data/ppm | Molecule number |
|---|---|---|
| 25.98 | 26.1 | 12 |
| 105.54 | 108.1 | 1 |
| 117.38 | 121.1 | 3 |
| 118.01 | 121.7 | 5 |
| 119.44 | 122.7 | 4 |
| 124.31 | 127.2 | 7 |
| 124.76 | 128.6 | 15 |
| 125.06 | 129.2 | 14 |
| 125.51 | 129.5 | 17 |
| 126.83 | 129.7 | 16 |
| 126.94 | 134.9 | 2 |
| 128.71 | 134.9 | 18 |
| 132.11 | 137.1 | 13 |
| 137.75 | 144.2 | 11 |
| 140.82 | 168.5 | 6 |
| 162.144 | 8 |
Anaylsis of the NMR Data
| Minor Product/ppm | Major Product/ppm |
|---|---|
| 123.73 | 124.31 |
| 138.28 | 137.75 |
The data predicted by the molecular mechanics data does match both experimental data for the 13C carbons NMR of the minor and major product. the data may be slightly less for each value but it does match up to experimental data. The molecular mechanics NMR may not match the data when the electron distribution affects the molecule itself, as the geometric isomer is not in a aromatic ring, the electron distribution will have little to no effect on the molecule itself.
In comparison with the NMR of the carbons in the olefin bond, it was found that at the carbon on the other side of the phenyl group, the trans Major product carbon is more deshielded and hence lies more to the the left of the NMR diagram than the carbon in the Cis Minor product. the difference is 0.58 ppm, which is a small but noticable change in the NMR of the cis and trans product. in the carbon on the olefin where the groups switches, minor one is now more deshielded than the trans carbon. in conclusion, though the NMR does show a very clear difference to which one is cis and trans, it would be far more better to do experimental data in order to see the even finer changes in the 13-C NMR between a cis or a trans olefin.
3J H-H coupling
In this product, the hydrogen on the olefin does not have a 3J coupling with any other hydrogen on either products and therefore the two products cannot be distingushed by the coupling constant. However, if there were two hydrogens on a 3J coupling, it would be possible to tell the difference as the trans product would have a larger coupling constant than the cis geometry.
IR and Vibrational Spectrum
As the products of both are just cis and trans to each other, the IR spectrum would be very near to each other and hence would not be useful in distingushing between the two different geometric isomers. The sum of the electronic and thermal free energies are also identical at -747.22 Hartree, and therefore will not help to distingush between the two geometric isomers.
References and Citations
- ↑ ="Regio- and stereoselective control in the addition of Grignard reagents to the pyridine ring system <DOI:10.1021/jo00356a016 "/
- ↑ ="Evaluation and prediction of the stability of bridgehead olefins <DOI:10.1021/ja00398a003 "/
- ↑ ="The type 2 intramolecular imino Diels-Alder reaction. Synthesis and structural characterization of bicyclo[n.3.1] bridgehead olefin/bridgehead lactams <DOI:10.1021/ja00059a022 "/
- ↑ ="Hyperstable olefins: further calculational explorations and predictions <DOI:10.1021/ja00274a016 "/
- ↑ =" Hydrogenation of [5]- and [6]Metacyclophane: Reactivity and Thermochemistry <DOI:10.1002/(SICI)1521-3765(20000502)6 "/
- ↑ =" A Reversal of pi-facial diastereoselection upon electronegative substitution of the substrate and the reagent ’’<DOI:10.1021/ja00204a018 "/
- ↑ =" A molecular orbital and crystallographic study of the structure and -facial regioselectivity of 9-chloro-1,4,5,8-tetrahydro-4a,8a-methanonaphthalene’’<DOI:10.1039/P29920000447 "/
- ↑ =" Palladium-Catalyzed Intramolecular 5-exo-dig Hydroarylations of N-Arylpropiolamides: Thermodynamics-Controlled Stereoselective Synthesis of 3-Methyleneoxindoles’’<DOI:10.1021/jo901963g "/
- ↑ " N.m.r. Spectra and Stereoisomerism in Pyrazolines’’<DOI:10.1021/jo01058a052 "/
- ↑ " Analysis of conjugated linoleic acid isomers by 13C NMR spectroscopy'’<DOI:10.1016/S0009-3084(98)00106-6 "/