
Jb406-1
The Hydrogenation of Cyclopentadiene Dimer
Comparisons of the computed energies of the potential pentadiene dimers 1
.gif)
and 2 revealed that dimer 2 was more stable than dimer 1, but only dimer 2 was observed experimentally. To investigate this I constructed both potential dimers in chemBIO 3D and examined their geometries and energies. Below are the tabulated energies from MM2 energy minimisation, and the structures of the 2 dimers.
.gif)
| Contribution (kcal) | ||
| Type of Energy | Dimer 1 | Dimer 2 |
| Stretch | 1.2864 | 1.2552 |
| Bend | 20.6030 | 20.8259 |
| Strech-bend | -0.8420 | -0.8336 |
| Torsion | 7.6545 | 9.5117 |
| Non-1,4-Van deWaals | -1.4327 | -1.4986 |
| 1,4-Van deWaals | 4.2351 | 4.3079 |
| Dipole-dipole | 0.3773 | 0.4452 |
| Total energy | 31.8816 | 34.0136 |
We can see from comparisons of the two potential dimers, 1 is more stable, but 2 is the only one seen. This is explained by looking at the mechanism of formation. The reaction is a electrocyclic reaction, with the total number of π electrons equal to 6. In π systems where no. electrons =4n+2 the transition state is of Huckel topology-the reactants have in phase π orbitals which twist suprafacially so that similar lobes face each other. The geometry of the orbitals would not proceed via this topology to produce dimer 1, only dimer 2 (under thermal conditions).


The hydrogenation of dimer 2 has 2 potential products, referred to here as 3 and 4. They differ in the double bond hydrogenated (as the dihydrogenated product is not produced without prolonged hydrogenation). Comparisons of the energies would suggest that 4 is the dominant product formed, while the geometry of the dimer with respect to angle of attack by protons suggests that there would be more steric hindrance for the formation of 4.
When we look at the energies of the two potential products, 4 is the more stable product (relative energies, 4 = 31.1767kcal, 3 = 35.9323kcal, determined by MM2 minimisation of energies).
- possible attack angles of hydrogenation upon the double bonds of dimer 2
-
 hindered by ring (attack leading to 3)
hindered by ring (attack leading to 3) -
 little to no hinderence (attack leading to 3)
little to no hinderence (attack leading to 3) -
 hindered by ring (attack leading to 4)
hindered by ring (attack leading to 4) -
 hindered by just 1 H (attack leading to 4)
hindered by just 1 H (attack leading to 4)




For both double bonds there are 2 possible general angles of attack; above the bond coming towards the existing hydrogen's, and below the bond coming towards the existing hydrogens. For the attack on the double bond leading to product 3, the attack angles are hindered by the ring structure from one direction, but from the other direction there is little to no hindrance due to the angle of attack (assumed to be ~109° from the plane of the double bond, opposite from the original hydrogen).
However with the attack leading to product 4 the attack angles are still hindered, but more so than with with product 3. There is still hindrance from the ring structure from one direction, but from the other direction there is a hydrogen, which would obviously have more of a steric hindrance than no hydrogen's from 3.
I would speculate that the relatively low hindrance combined with the lower total energy would favour 4 as the dominant product of hydrogenation under thermodynamic conditions, but under kinetic conditions, I would expect 3 to be more dominant.
However without actual experimental data all this is is speculation; we need actual experimental data to draw any conclusions.
Stereochemistry of Nucleophilic additions to a pyridinium ring (NAD+ analogue)


For this part of the module I was investigating the source of the stereocontrol within reactions. The reaction summaries are to the right. I constructed the rings within chemBIO 3D and examined their 3D shape to see whether the stereocontrol could be infered from this.
Images: taken from guide[1]

In the first reaction shown within the subsection(5 to 6), a MM2 optimisation was performed on the starting reagent. (prolinol) I tried several different initial conformations before performing MM2 optimisation, and from the lowest energy optimisation and from further analysis of this lowest energy conformation within chemBIO 3D, represented in the thumbnail to the left, I inferred that the reaction proceeded as it does because the oxygen lies above the plane of the aromatic ring.
This would affect the stereocontrol, since the Mg would co-ordinate to the oxygen. Here the free roation of the the methyl group attached to the Mg atom would be limited due to steric hindrance, which would prevent the Me from assuming position anywhere else but above the aromatic ring (the hindrance being from hydrogen's on the other side, and the iodine also attached to the Mg being repulsed from the highly negative aromatic ring).

In the second reaction (7 to 8) shown again we can see that within the reagent (pyridinium) the carbonyl group lies above the aromatic ring, as demonstrated in this diagram. This again will mean that the incoming molecule will bond to this, placing the nitrogen above the aromatic ring, preventing it from attacking from any other position, giving the resultant geometry.
References
---
Stereochemistry and Reactivity of an Intermediate in the Synthesis of Taxol

Comparision of the two isomers within chemview3D showed that isomer 10 was the more stable of the two possible( 51.7961 kcal/mol, compared with 60.4776 kcal/mol for isomer 11), suggesting that 11 converted to 10 over time (a thermodynamically driven reaction).
Further stabilisation which was not automatically added by the program is the chair conformer of the cylcobutene ring, though manual manipulation and resultant minimisation of energy by MM2 showed this to be indeed a more stable structure for the molecule (molecules energy is then 47.3580 kcal/mol).
Suggested in the module 1 guide is that the reduced reactivity in the resultant intermediate is due to the fact it is a hyperstabilised alkene. Futher reading showed that this was a phenomenon where cycloalkenes are more stable than there respective cycloalkenes, due to to transannular and vicinal hydrogen interactions. [1]
This would correspond to the observed 3D structure of the taxol intermediate; the geometry of the molecule leaves the carbonyl group in an optimal position to interact with the hydrogens. Reducing the double bond destroys the unique geometry that allows these interactions to occur, so raising the energy of the molecule.
Images:Taken from guide[2]
References
- ↑ Tetrahedron, Vol. 53, No. 28, pp9727-9734, 1997
- ↑ http://www.ch.ic.ac.uk/wiki/index.php/Mod:organic
How one might induce room temperature hydrolysis of a peptide

In this section I investigate the observed kinetic behaviour of 2 peptide hydrolsis reactions by computational analysis of the two molecules. The two peptides are refered to as 13 and 14, and the reaction schematic is to the right, with their respective half lifes posted with them.
- All isomers of all peptides
-
 Peptide 13 equitorial isomer
Peptide 13 equitorial isomer -
 Peptide 13 axial isomer
Peptide 13 axial isomer -
 Peptide 14 axial isomer
Peptide 14 axial isomer -
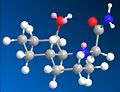 Peptide 14 equitorial isomer
Peptide 14 equitorial isomer




Examining the two posibble peptides and their possible isomers, and determining their optimum geometry with MM2 it becomes obvious that the isomer of 13 where the N substituent is axial (all isomers refered to in respect to the orientation of this group from now on) is the only one where a potential reaction is possible, once the molecules have had their orientation optimised. The equitorial isomers orientation is so that the reactive amine group is facing away from the carbonyl group.
With 14 and its isomers it becomes obvious that neither has an optimal geometry for the reaction - in both conformations the alcohol group is stabilly hydrogen bonded to the carbonyl substituent on the N substituent - or so MM2 would imply. The rotation required for the bond to rotate so that the amide group and the alcohol group could react would be highly destabilising in breaking the hydrogen bond-a probable reason why the half life for this reaction is so large.
Images taken from guide[1]
References
Regioselective Addition of Dichlorocarbene

In this section of this module I used more accurate geometry calculations to determine the structure and bond lengths of molecules. The molecule that i was investigating is represented to the right, and I was using the data gained to make predictions and explain observed behaviour.
Here I optimised the geometry with MM2 first, then ran a calculation to provide a MO representation, then used a more accurate density functional approach.
I then compared compound 12 with its 2 potential partially hydrogenated versions (the double bond that is hydrogenated being different in each case, represented to the left). I then optimised them and examined the differences between the vibrations and bond lengths between all versions.
- Energy levels of unhydrogenated product
-
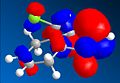 HOMO-1
HOMO-1 -
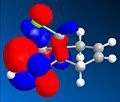 HOMO
HOMO -
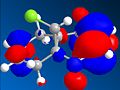 LUMO
LUMO -
 LUMO + 1
LUMO + 1 -
 LUMO + 2
LUMO + 2





From analysis of the structure of the HOMO, LUMO, HOMO-1, LUMO+1 and LUMO+2, as well as the optimised bond lengths in gaussviews I was able to draw several conclusions. Firstly that the double bond anti the chloro is less suceptable to electrophilic attack. This is due to the fact that the π orbitals contribute to the C-Cl σ* orbital, weakening the double bond (shown by the lengthening of it) and reducing the lectron density over the double bond. This is supported by looking at the difference between the C-Cl bond lengths - it is longer, showing stabilisation, with the double bond that is anti present. This is explained by looking at the LUMO and HOMO diagrams- the HOMO shows the postion of the π orbitals, while the LUMO+2 shows the position of the C-Cl σ* orbital. If we compare the two we can see that either way the phases of the σ* and the π orbitals match up, which would encourage the formation of the LUMO+2 fomration, with inividual charges based around the C and the Cl, suggesting that this promotes the breakdown of a covalent bond and the promation of a ionic bond, leading to a closer bond.
All the bond lengths were taken from analysis in gaussview; though analysis of the IR spectrum showed the wavelength of the C-Cl bond shifted to a longer wavelength, showing that the bond had lengthened, though as this was also calulated via a computational method this is not a reliable check, as this data would have been obtained via the same method.
Structure based Mini project using DFT-based Molecular orbital methods
In this part of the module I examined a reaction with 2 possible stereochemically distinct products, which had already been examined in literature[1], and constructed them in chemBIO 3D to see whether the predited results matched the observed litrature information[2].
I chose to examine the assigning of regioisomers in "click chemistry" - more specifically the reaction between an azide and an alkyne to give a 1,2,3-triazole (a 1,3-dipolar cycloaddition). There are 2 possible products to this reaction, depending on the catalyst used, with the difference in geometry being the place where the the substituent of the alkyne is positioned.
To fully investigate this I took a molecule examined in the literature, number 1, where the substituient of the alkyne is a benzene ring, and the same again attached to the azide (see diagram right).
Both potential molecules geometry was optimised, and then the 13C NMR, IR spectrum and optical rotation of the 2 potential products predicted. These were chosen since they are the most common non destructive techneches, and the only ones covered in this course.
Comparision of the predicted 13C NMR
- Predicted and actual NMR of isomers A and B
-
 13C predicted NMR of A
13C predicted NMR of A -
 13C predicted NMR of B
13C predicted NMR of B -
![Actual 13C NMR of A[3]](/images/thumb/d/d6/13CNMRA.jpg/120px-13CNMRA.jpg) Actual 13C NMR of A[3]
Actual 13C NMR of A[3] -
![Actual 13C NMR of B[4]](/images/thumb/c/ca/13CNMRB.jpg/120px-13CNMRB.jpg) Actual 13C NMR of B[4]
Actual 13C NMR of B[4]
![Actual 13C NMR of A[3]](/wiki/File:13CNMRA.jpg)
![Actual 13C NMR of B[4]](/wiki/File:13CNMRB.jpg)
For the NMR of A there is overlap of the 2 phenol rings. The carbon linking the bezene ring to the triazole is very deshielded, and the carbon adjacent to it is more shielded, while the carbon linking to the phenol ring is most shielded.
When we look at the 13C NMR of B, again there is overlap of the phenol rings, but the carbon that was part of the triazole was overlaped with the carbon that was liked in the phenol ring, and both were deshielded from the overlap of the phenol rings. Again the carbon in the triazole that links the benzene ring was the most deshielded, and the carbon joining the phenol ring to the triazole ring was the most shielded. There was tighter grouping of the 13C NMR here than in the NMR of A.
Looking at the 2 predicted NMR would suggest that they would be sufficiently distict to be determined by 13C NMR, but these are just the predicted NMR. To actually see whether this is a suitable method, we have to compare it to the actual NMR, referenced in the original litrature documentation.
When we look at the actual NMR we see that indeed that there is overlap of the 2 phenol rings, but they have been shifted to a different shielding. However the 2 sets of NMR are distinct; in both the predicted and the actual the NMR of A was more diffuse, though some of the peaks were different. The most shielded and deshielded peaks were in the same positions.
IR Analysis
- predicted IR spectra
-
 predicted IR spectrum of A
predicted IR spectrum of A -
 predicted IR spectrum of B
predicted IR spectrum of B


I also used the computational programs to investigate the potential IR of the isomers - they were very similar spectra, though the intensity of the some peaks were higher in one or the other, at ~750cm-1 for B and ~3200cm-1 for A. The stronger stretch for B refers to a entire molecule strrtch, while the strech for A is just the concerted hydrogen streching on the bezene directly off the triazole ring. This would suggest that you coyuld use the IR to differentiate between the 2, but without actual experimetal data the veracity of this prediction cannot be known(no such data was provided with the litrature).
Conclusion
Looking at both sets of predicted data I would say that the 13C NMR would be the most reliable way to differentiate between the 2 isomers, as there are distinct lines and patterns present in the spectra for both isomers, while the IR of the 2 molecues was incredibally similar, with only intensity differentiating the 2, but this would affected by external factors.
- ↑ http://pubs.acs.org/doi/abs/10.1021/ja054114s
- ↑ http://pubs.acs.org/doi/suppl/10.1021/ja054114s/suppl_file/ja054114ssi20051014_012328.pdf
- ↑ http://pubs.acs.org/doi/suppl/10.1021/ja054114s/suppl_file/ja054114ssi20051014_012328.pdf
- ↑ http://pubs.acs.org/doi/suppl/10.1021/ja054114s/suppl_file/ja054114ssi20051014_012328.pdf