
IMM2:01352784
Introduction to Molecular Modelling 2
Optimization
NH3
NH3 |
- Calculation Method : RB3LYP
- Basis Set : 6-31G(d,p)
- Final Energy E(RB3LYP) : -56.55776873 a.u.
- RMS Gradient Norm : 0.00000485 a.u.
- Point Group : C3v
- N-H distance : 1.01798 Å
- H-N-H angle: 37.129°
- Gaussian Final Optimization:
Item Value Threshold Converged?
Maximum Force 0.000004 0.000450 YES
RMS Force 0.000004 0.000300 YES
Maximum Displacement 0.000072 0.001800 YES
RMS Displacement 0.000035 0.001200 YES
Predicted change in Energy=-5.986265D-10
Optimization completed.
-- Stationary point found.
----------------------------
! Optimized Parameters !
! (Angstroms and Degrees) !
-------------------------- --------------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------------------
! R1 R(1,2) 1.018 -DE/DX = 0.0 !
! R2 R(1,3) 1.018 -DE/DX = 0.0 !
! R3 R(1,4) 1.018 -DE/DX = 0.0 !
! A1 A(2,1,3) 105.7412 -DE/DX = 0.0 !
! A2 A(2,1,4) 105.7412 -DE/DX = 0.0 !
! A3 A(3,1,4) 105.7412 -DE/DX = 0.0 !
! D1 D(2,1,4,3) -111.8571 -DE/DX = 0.0 !
--------------------------------------------------------------------------------
GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
Vibration Analysis

- Modes Expected (3N-6) : 6
- Degenerate Modes : 2,3 and 5,6
- Bending Vibration modes : 1 (Symmetric Bending), 2 (Asymmetric Bending) and 3 (Asymmetric Bending, scissoring)
- Stretching Vibration modes : 4 (Symmetric Stretching), 5 (Asymmetric Stretching) and 6 (Asymmetric Stretching)
- Mode 4 is highly symmetric
- Mode 1 is the 'umbrella mode' (Symmetric Bending)
- There should be 4 bands in an experimental spectrum of gaseous ammonia
- There are no negative frequencies, molecule is optimized correctly
Charge Analysis

Partial Charge on N : -1.125 Partial Charge on H : 0.375
N holds a negative partial charge as it is more electronegative than H. The partial charge on N is large however, holding more than a full negative charge with -1.125 e. The sum of the partial charges correctly equals to 0 as it should be for a neutral molecule.
H2
H2 |
- Calculation Method : RB3LYP
- Basis Set : 6-31G(d,p)
- Final Energy E(RB3LYP) : -1.17853930 a.u.
- RMS Gradient Norm : 0.00012170 a.u.
- Point Group : D∞h
- H-H distance : 0.74309 Å
- H-H angle: 180°
- Gaussian Final Optimization:
Item Value Threshold Converged?
Maximum Force 0.000211 0.000450 YES
RMS Force 0.000211 0.000300 YES
Maximum Displacement 0.000278 0.001800 YES
RMS Displacement 0.000393 0.001200 YES
Predicted change in Energy=-5.852867D-08
Optimization completed.
-- Stationary point found.
----------------------------
! Optimized Parameters !
! (Angstroms and Degrees) !
-------------------------- --------------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------------------
! R1 R(1,2) 0.7431 -DE/DX = -0.0002 !
--------------------------------------------------------------------------------
GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
Vibration Analysis

- There is only 1 symmetric stretching vibration mode, being a linear molecule
- There are no negative frequencies, molecule is optimized correctly
N2
N2 |
- Calculation Method : RB3LYP
- Basis Set : 6-31G(d,p)
- Final Energy E(RB3LYP) : -109.52412865 a.u.
- RMS Gradient Norm : 0.00015191 a.u.
- Point Group : D∞h
- N-N distance : 1.10541 Å
- N-N angle: 180°
- Gaussian Final Optimization:
Item Value Threshold Converged?
Maximum Force 0.000263 0.000450 YES
RMS Force 0.000263 0.000300 YES
Maximum Displacement 0.000082 0.001800 YES
RMS Displacement 0.000116 0.001200 YES
Predicted change in Energy=-2.162123D-08
Optimization completed.
-- Stationary point found.
----------------------------
! Optimized Parameters !
! (Angstroms and Degrees) !
-------------------------- --------------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------------------
! R1 R(1,2) 1.1054 -DE/DX = 0.0003 !
--------------------------------------------------------------------------------
GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
Vibration Analysis

- There is only a single symmetric stretching vibration mode as it is a linear molecule.
- There are no negative frequencies, molecule is optimized correctly
Reaction energy of N2 + 3H2 -> 2NH3
Using Hess's law, the reaction energy can be found by calculating the sum of energy of the product minus the sum of energy of the reactants
- E(NH3)= -56.55776873 a.u.
- 2*E(NH3)= -113.11553746 a.u.
- E(N2)= -109.52412865 a.u.
- E(H2)= -1.17853930 a.u.
- 3*E(H2)= -3.53561790 a.u.
- ΔE=2*E(NH3)-[E(N2)+3*E(H2)]= -0.05579091 a.u.
- Conversion of ΔE to kJ/mol : -0.05579091*2625.5 / 2 = -73.2395171025 kJ/mol
As the reaction is exothermic, work has been given out by the reaction and the products will have less ability to do work in the future. i.e. the products (NH3) is more stable.
Optimisation of CN-
CN- |
- Calculation Method : RB3LYP
- Basis Set : 6-31G(d,p)
- Final Energy E(RB3LYP) : -92.58147665 a.u.
- RMS Gradient Norm : 0.126373541 a.u.
- Point Group : C∞h
- C-N distance : 1.8408 Å
- C-N angle: 180°
- Gaussian Final Optimisation:
Item Value Threshold Converged?
Maximum Force 0.000000 0.000450 YES
RMS Force 0.000000 0.000300 YES
Maximum Displacement 0.000000 0.001800 YES
RMS Displacement 0.000000 0.001200 YES
Predicted change in Energy=-1.376128D-14
Optimization completed.
-- Stationary point found.
----------------------------
! Optimized Parameters !
! (Angstroms and Degrees) !
-------------------------- --------------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------------------
! R1 R(1,2) 1.1841 -DE/DX = 0.0 !
--------------------------------------------------------------------------------
GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
Vibration Analysis


- There is only a single symmetric stretching vibration mode as it is a linear molecule, and this involves a change in dipole. Hence there is only one band in the experimental spectrum.
- Expected number of modes (3N-5) is 1, which is as predicted.
- There are no negative frequencies, molecule is optimized correctly
Charge Analysis

- There is a negative charge on the CN- ion, however it is distributed unevenly with -0.246 on C and -0.754 on the N. The N atom carries more of the negative charge due to the fact that it is more electronegative than C. Since the CN- ion carries a -1 charge, the sum of the values equal -1.
MO Analysis


- The orbitals seen here are the 1s orbitals of both N and C respectively. Both orbitals are very deep in energy with -14.00393 au of N and -9.86720 au in C. It is much deeper in energy in N due to the increase in effective nuclear charge. Because they are so deep in energy and buried deep inside the atom, they are unable to interact with the other orbitals, even between each other as the difference in energy is 4.13673 au, which is large enough that they do not interact. Hence the orbitals only surround the individual atoms they belong to.

- This occupied bonding molecular orbital is a result of the interaction between the 2s atomic orbitals of both N and C when both are in the in phase as each other. This bonding orbital is the deepest in energy of all molecular orbitals at -0.56195 au, being in the HOMO region. This MO contributes to the sigma bonding between N and C.The 2s orbital energy in N is closer to this bonding orbital than C, so the molecular orbital lobe is larger around N than C. The 2s orbital energy in N is closer to this bonding orbital than C, so the molecular orbital lobe is larger around N than C.

- This occupied sigma anti-bonding molecular orbital comes from the interaction of the 2s atomic orbitals of N and C when they are out of phase of each other. This is higher in energy than the sigma bonding orbital previously mentioned. The orbital is still within the HOMO region and has a node in between N and C. As the 2s orbital energy in C is slightly closer to this anti bonding orbital than the 2s orbital in nitrogen, the molecular orbital lobe on the C atom is slightly larger than the one found on N.

- This occupied bonding molecular orbital comes from the interaction between the 2pz orbitals in both N and C when they are in phase. There is a node on each atom respective as a result of being constructed out of the 2pz orbitals. This is the HOMO, the energy of this orbital is valued at 0.01857 au. This contributes to the sigma bonding between N and C.
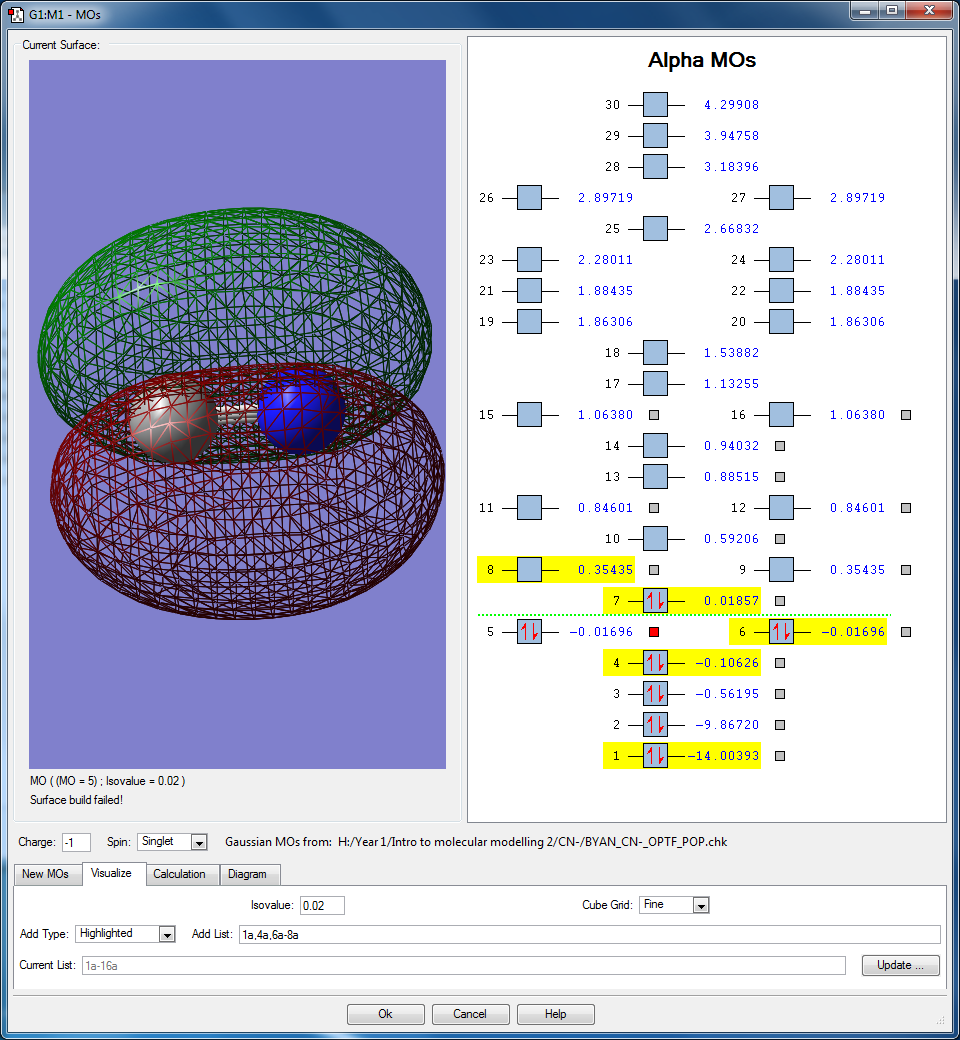
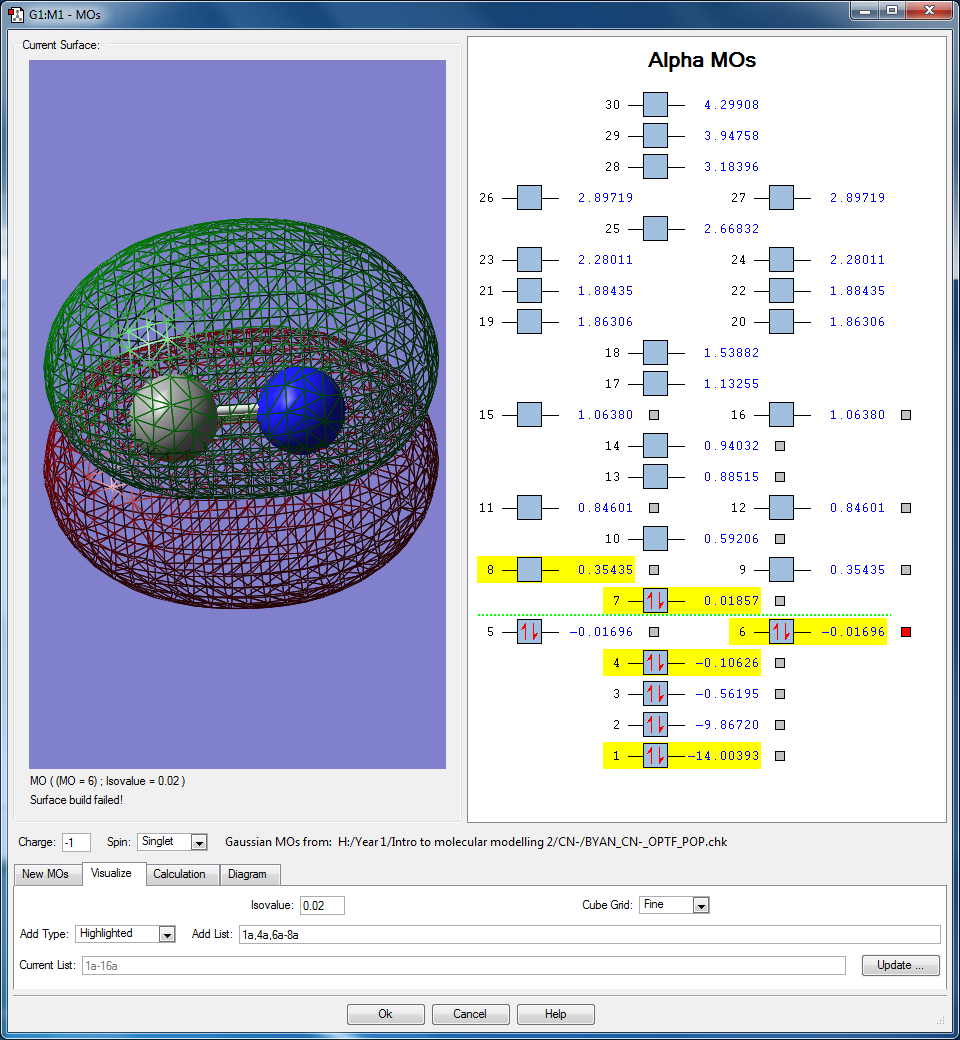


- Here are 2 degenerate occupied bonding molecular orbitals that originate from the interaction between the 2px and 2py atomic orbitals found in both N and C, thus the orbitals are orthogonal to each other. The molecular orbital lobes are found above and below the C-N bond. Their energies are in between the HOMO and the sigma anti bonding orbital, at -0.01696 au. These two orbitals contributes to the pi bonding orbitals found in the molecule.
Optimisation of H2SiO
H2SiO |
- Calculation Method : RB3LYP
- Basis Set : 6-31G(d,p)
- Final Energy E(RB3LYP) : -365.90001403 a.u.
- RMS Gradient Norm : 0.00000941 a.u.
- Point Group : C2v
- Si-H distance : 1.48652 Å
- Si-O distance : 1.53172 Å
- O-Si-H angle: 124.157°
- H-Si-H angle: 111.686°
- Gaussian Final Optimisation:
Item Value Threshold Converged?
Maximum Force 0.000023 0.000450 YES
RMS Force 0.000009 0.000300 YES
Maximum Displacement 0.000022 0.001800 YES
RMS Displacement 0.000015 0.001200 YES
Predicted change in Energy=-4.981733D-10
Optimization completed.
-- Stationary point found.
----------------------------
! Optimized Parameters !
! (Angstroms and Degrees) !
-------------------------- --------------------------
! Name Definition Value Derivative Info. !
--------------------------------------------------------------------------------
! R1 R(1,2) 1.4865 -DE/DX = 0.0 !
! R2 R(1,3) 1.4865 -DE/DX = 0.0 !
! R3 R(1,4) 1.5317 -DE/DX = 0.0 !
! A1 A(2,1,3) 111.6859 -DE/DX = 0.0 !
! A2 A(2,1,4) 124.1571 -DE/DX = 0.0 !
! A3 A(3,1,4) 124.1571 -DE/DX = 0.0 !
! D1 D(2,1,4,3) 180.0 -DE/DX = 0.0 !
--------------------------------------------------------------------------------
GradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGradGrad
H2SiO Gaussian Optimization Log
Vibration analysis

- The number of modes expected (3N-6) is 6.
- Mode 1 : out of plane wagging
- Mode 2 : In plane rocking
- Mode 3 : In plane scissoring
- Mode 4 : In plane scissoring of the H atoms and stretching of the O atom
- Mode 5 : Symmetric stretching of H atoms
- Mode 6 : Asymmetric stretching of H atoms
- All of the vibrations involves a change in dipole moments, so all vibrations are IR active, hence 6 bands are expected in an experimental spectrum.
- There are no negative frequencies, so molecule is optimized correctly.
Charge analysis

H2SiO is overall a neutral molecule, however due to the differences in electronegativity, partial charges are present. Especially between Si and O as O is a lot more electronegative than both Si and H, hence picking up most of the negative partial charge at -1.001 e, almost practically a full negative charge on the O atom. Si being the least electronegative atom in the whole molecular, will pick up a partial positive charge as expected at 1.472 e, this is a large 1.5 e of positive charge on Si atom. H atoms being more electronegative than Si but less than O is in the middle in terms of partial negative charge at -0.236 e. The sum of these charges correctly equal 0, as it should be for a neutral molecule. Due to the large charge separation between O and Si, the molecule is expected to be extremely unstable.