
3rd Year Computational Chemistry: Module 2 (gc2109)
By Gen Cross
This module aims to illustrate the ability of computational chemistry to predict the structure and relative energies of various inorganic molecules. In addition to predict the IR spectra and Molcular Orbital structures of these optimized structures. Computaional techniques are useful for analyzing unusual bonding systems which are more common in inorganic chemistry, and particulary in cases where analyses is either extremely difficult in a wet lab (e.g. very unstable small moecules) or very toxic compounds.
Throughout this module the Gaussian 09W programme is used to carry out the calculations on the molecules, and GaussView 5.0 is used to draw structures and create input files. These input files are sent to the HPC server for calculation requiring more computational power. The output files gathered from these calcuations are then analysed using GaussView 5.0 to visulise optimizeds structures, bond vibrations and molecular orbitals. During these geometry optimizations, there are generally two main parts the SCF and OPT calculation, dealing with electrons and nuclei respectively. First the Schrodinger equation is solved for the electrons in the molecules using the Born-Oppenheimer approximation. Then the positions of the nuclei are optimized, and the process is repeated until a point at which the gradient of the energy = 0. Generally a minimum point is being searched for as this corresponds to the equilibrium bond lengths for the molecule (there are no forces pushing or pulling the nuclei). However the gradient also = 0 at a maximum point corresponding to a transition state, therefore the second derivative is alse found, when positive this confirms a minium point has been found and the optimal geometry has been found.
Bonding (ab intio and density functional molecular orbitals)
BH3
Introduction
This sections aims to show that computational methods can be used to optimize the geometry of a simple inorganic molecule such as BH3 and once this geometry is found how it can be analysed to predict IR absorptions and the location of molecular and non-bonding orbitals.
Geometry Optimisation of BH3
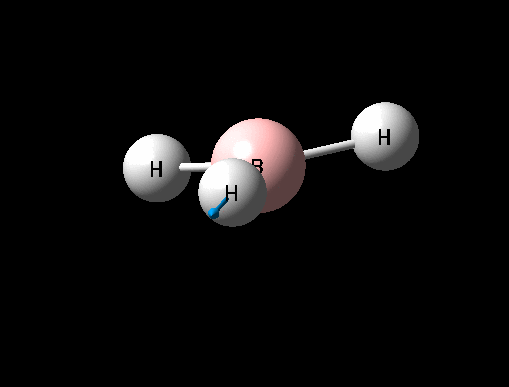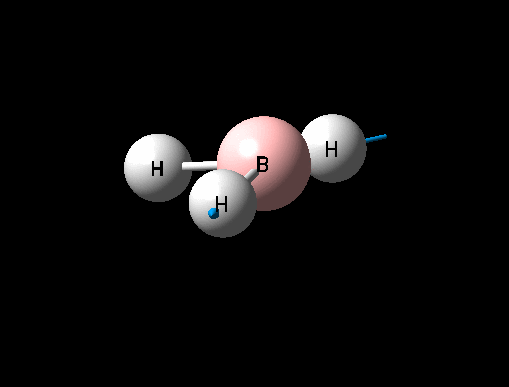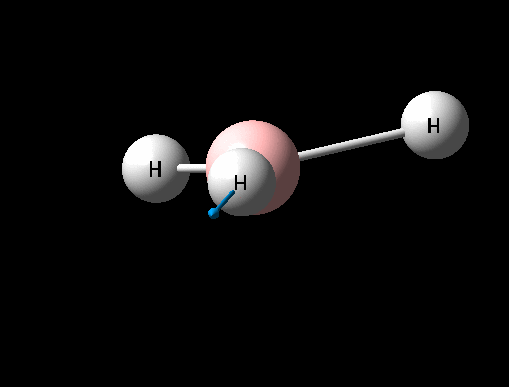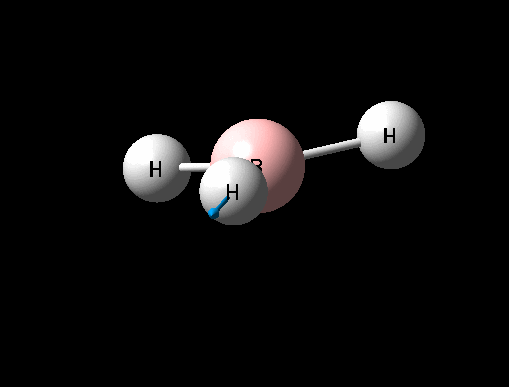
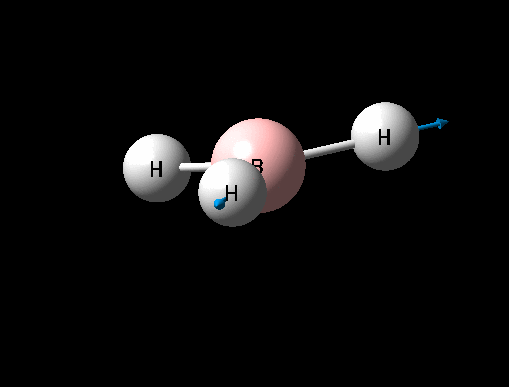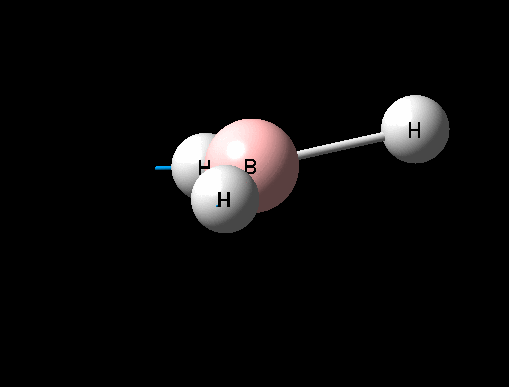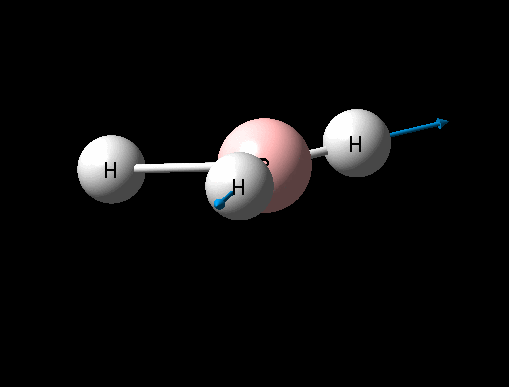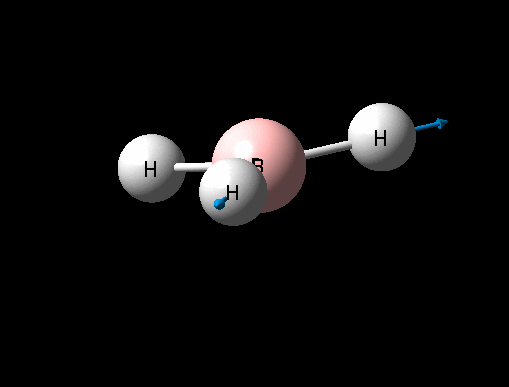
A BH3 was first drawn on GuassView 5.0 with a trigonal planar structure. Each of the B-H bonds lengths were then set to 1.5 Angstroms. This molecule was optimized using the 'Guassian Calculation Setup' function to submit the molecule to Guassian 09W on the computer, with the B3LYP method and the 3-21G basis set. The advantages of this was that it was a quick calculation since the basis set is small, the disadvantage is that the some accuracy maybe lost. On such a simple molecule this inaccuracy will be small so the use of such a basis set is justified. The 3D view of the optimised BH3 molecule can be seen here .
The more relevant information from the LOG FILE is shown below, but it has also been published in full detail here.

Optimized Parameters
(Angstroms and Degrees)
------------------- ---------------------
Name Definition Value Derivative Info.
---------------------------------------------------------------
R1 R(1,2) 1.1935 -DE/DX =0.0004
R2 R(1,3) 1.1935 -DE/DX =0.0004
R3 R(1,4) 1.1935 -DE/DX =0.0004
A1 A(2,1,3) 120.0 -DE/DX =0.0
A2 A(2,1,4) 120.0 -DE/DX =0.0
A3 A(3,1,4) 120.0 -DE/DX =0.0
D1 D(2,1,4,3) 180.0 -DE/DX =0.0
---------------------------------------------------------------
Item Value Threshold Converged?
Maximum Force 0.000413 0.000450 YES
RMS Force 0.000271 0.000300 YES
Maximum Displacement 0.001610 0.001800 YES
RMS Displacement 0.001054 0.001200 YES
Predicted change in Energy=-1.071764D-06
Optimization completed.
-- Stationary point found.
It can be seen from the above that the optimization was successful, indicated by the value for the Root Mean Squared of the graph being very close to 0. This is confirmed by Gaussian saying the calculation has converged. It can be seen from the graphs below that the optimisation was complete in 4 iterations.


It can be seen from the data in the 'Optimisation Parameters' section and the Jmol diagram that the fully optimised structure for BH3 has B-H bond lengths of 1.19Å and bond angles of 120 degrees. This agrees well with literature. It can also be seen that the dipole moment of BH3 is 0 Debye, this is due to its high symmetry point group (D3H).
Frequency Analysis

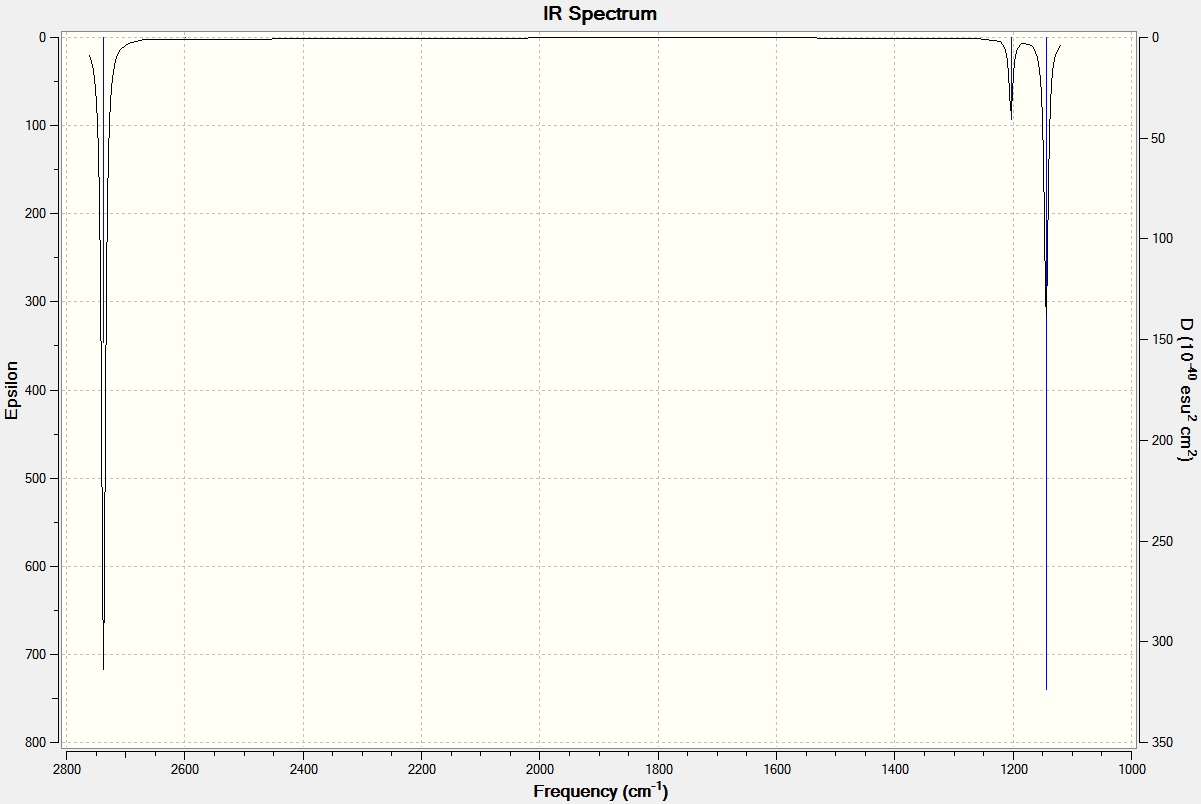
The purposes of carrying out frequency analyses on the optimised structure are 2 fold. First clearly a useful prediction of IR stretches and bond absorptions can be made, to compare with literature from the wet lab. Second, the vibrational analyses is effectively used to check the second derivative of the energy graph, i.e. to check if the stationary point found in the geometry optimisation is indeed a global minimum or is in fact a maximum corresponding to a transition state. Maxima are indicated by negative frequencies in the IR spectrum. It can be seen from the LOG FILE and the results below that the optimisation carried out above was successful.
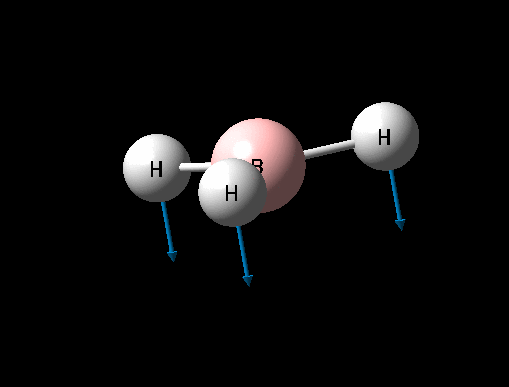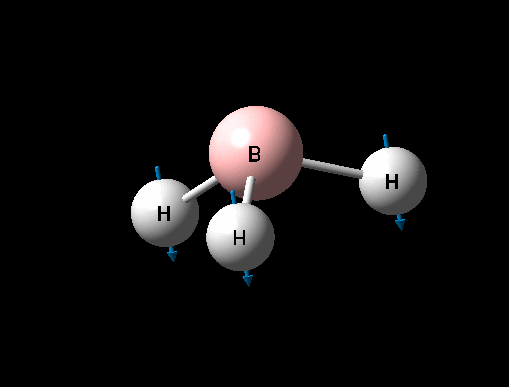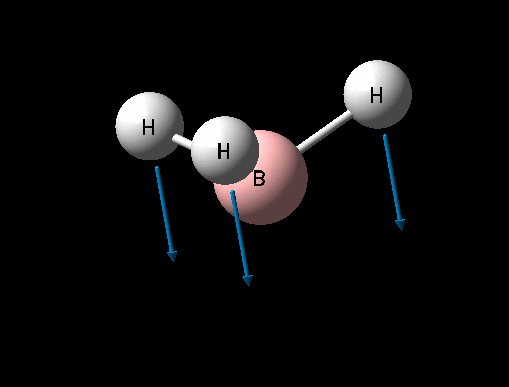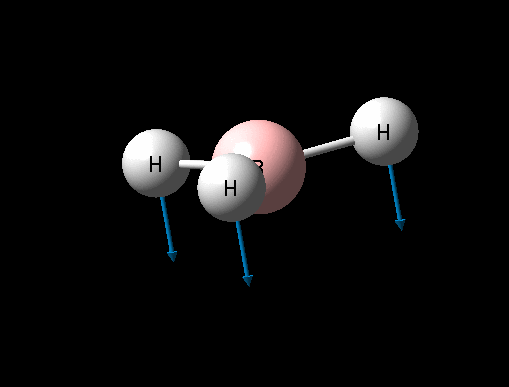
| Description for Mode of Vibration | Type of Vibrational Mode | Intensity of Absorption | Frequency of Absorption (cm-1) | Literature value for Frequency (cm-1)[1] | Percentage Error (%) | Diagram | Link to 3D animation |
|---|---|---|---|---|---|---|---|
| Umbrella deformation | A2 " | 93 | 1146 | 1225 | 6 |  |
Show Animation |
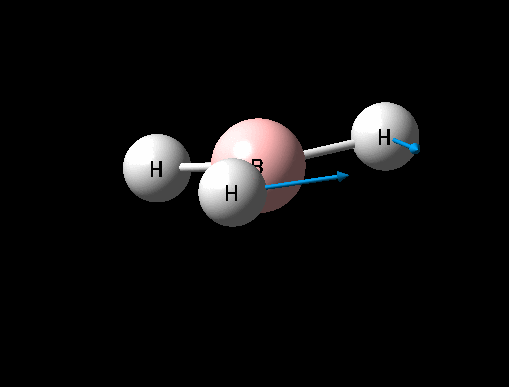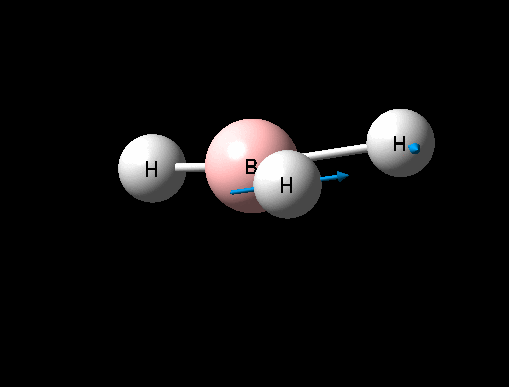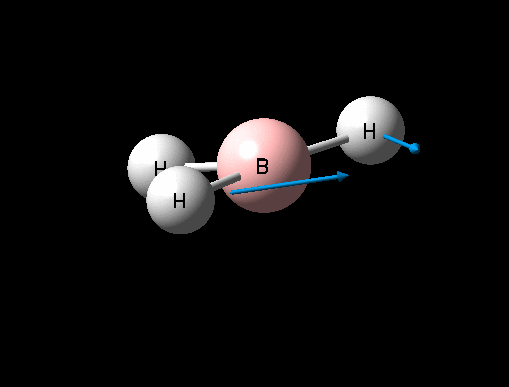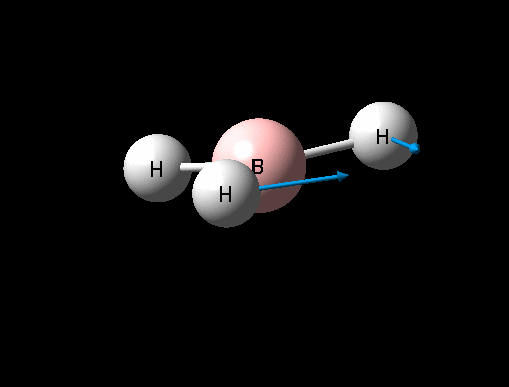
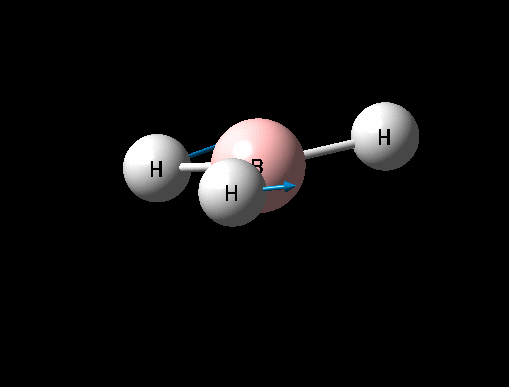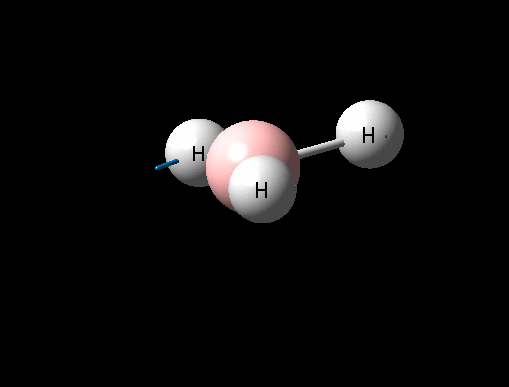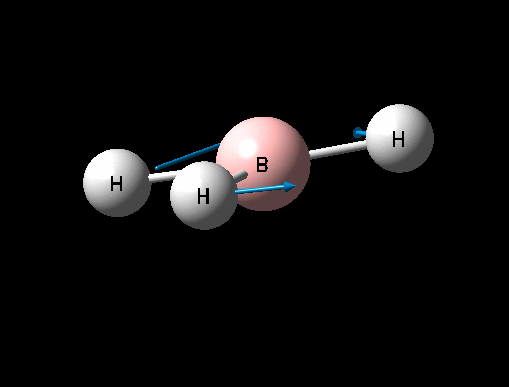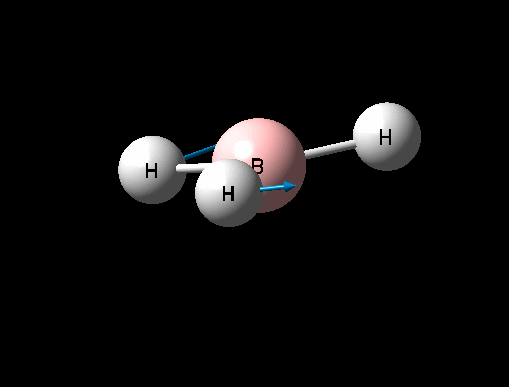
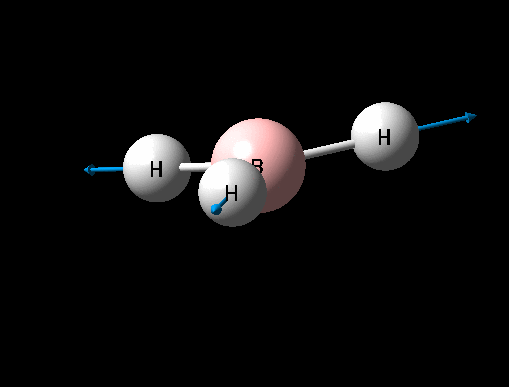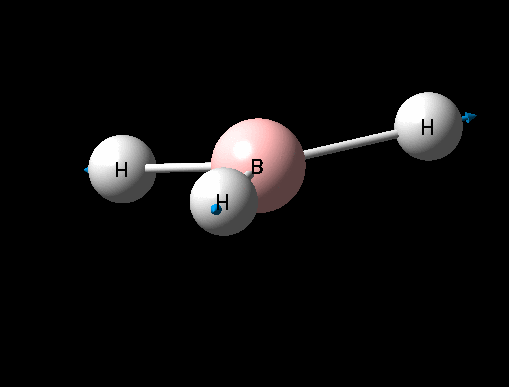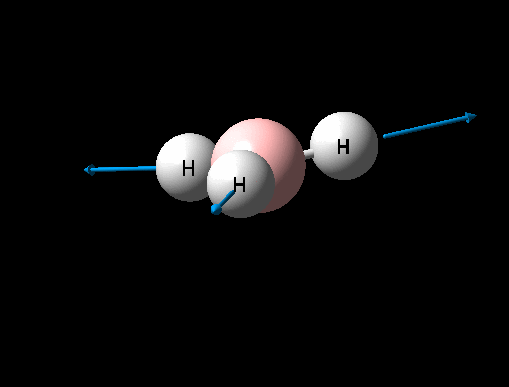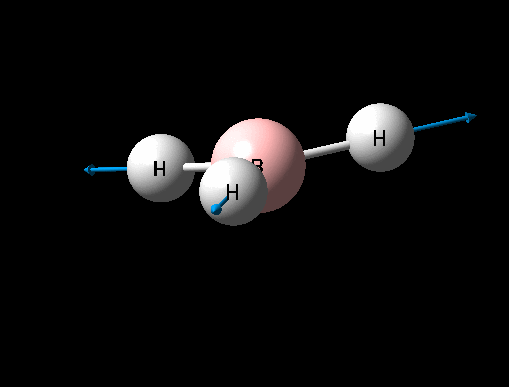
| Symmetric B-H scissoring | E' | 12 | 1205 | 1305 | 8 |  |
Show Animation |
| Asymmetric B-H rock | E' | 12 | 1205 | 1305 | 8 |  |
Show Animation |
| Symmetric B-H stretch | A1' | 0 | 2592 | - | - |  |
Show Animation |
| Asymmetric B-H stretch (1) | E' | 104 | 2730 | 2693 | 1 |  |
Show Animation |
| Asymmetric B-H strech (2) | E' | 104 | 2730 | 2813 | 3 |  |
Show Animation |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
It can seen from the data in the table above that there are 6 vibrational modes for BH3, however in the IR spectrum only 3 absorptions are seen. The 3 missing peaks can be explained by looking at the absorption intensities and their frequencies. It can be seen that the A' B-H stretch has an intensity of 0 because it is a totally symmetric stretch and therefore there is no change in overall dipole moment, an absorption is therefore not seen. In addition each pair of E' absorptions at 1205cm-1 and 2730cm-1 are exactly degenerate and therefore their absorptions overlap.
It should be noted that the literature values quoted in the table are themselves computational calculations by DeFrees and McClean using HF/6-31G*. That being said a good agreement with the literature is shown.
Analysis of the low frequencies suggests the vibrational results are accurate since they are very close to zero.
Full mass-weighted force constant matrix: Low frequencies --- -0.3716 -0.0098 -0.0018 37.2444 37.9574 37.9592 Low frequencies --- 1146.0292 1204.8631 1204.8641
Molecular Orbital Analysis
In order to find the molecular orbitals for BH3 the previously optimised structure was sent to the HPC server for an Energy analyses with the parameters 'pop=full' and 'full nbo' selected in the calculation set up. Once the calculation was complete the Formatted Checkpoint File was downloaded and the MOs visualised in GaussView 5.0. The LOG file can be seen here [2].
To check the validity of the quantitative MOs produced computational a molecular orbital diagram was predicted qualitatively using the linear combination of atomic orbitals method. This is shown below.
| MO Diagram with Orbitals shown in the XY plane | Orbitals seen in XZ Plane | Molecular Orbital |
|---|---|---|
 | ||
 |
LUMO+3 | |
 |
LUMO+2 | |
 |
LUMO+1 | |
 |
LUMO | |
 |
HOMO | |
 |
HOMO-1 | |
 |
HOMO-2 | |
 |
HOMO-3 |
The MO diagram above was produced using ChemDraw Pro 12.0
Hydrogen (2.20) [3] is slightly more electronegative than Boron (2.04) and so the H3 fragment was placed lower in MO diagram. Each of the fragment orbitals were assigned a symmetry group in order to ascertain which orbitals could mix. Finally the electrons were added to the MO diagram. The predicted structure for the MO diagram agrees well with the energies of the MOs calculated computationally so they were added into the figure above. It can be seen that the Boron 1s atomic orbital has been included in the diagram but is not actually involved in bonding as it is too low lying, backed up by the very low energy (-6.73 Ha) relative to the other MOs.
Qualitatively the order of the 2 highest energy MOs is difficult to predict since it can be argued that either should be lower in energy, on the one hand clearly the a1' fragment orbitals are lower in energy originally so it could be argued the resultant 3a' MO should also be lower in energy. Conversely the e' fragment orbitals show worse overlap and so it could be argued this interaction should be smaller leading it to be lower energy MO. In the end the diagram was drawn to agree with the energies found computationally.
Overall it can be seen that the ability of computational methods to predict the MOs for small molecules such as BH3 is very good. This accuracy would decrease and also require far more computational power and larger basis sets as more complex molecules are analysed.
NBO Analysis
The same LOG file [4] produced by energy calculation can be used for Natural Bond Order (NBO) analysis because the 'full NBO' option was added previously. The pertinent data from the LOG file can be seen below, and the full version is available here.

Summary of Natural Population Analysis:
Natural Population
Natural -----------------------------------------------
-----------------------------------------------------------------------
B 1 0.33161 1.99903 2.66935 0.00000 4.66839
H 2 -0.11054 0.00000 1.11021 0.00032 1.11054
H 3 -0.11054 0.00000 1.11021 0.00032 1.11054
H 4 -0.11054 0.00000 1.11021 0.00032 1.11054
=======================================================================
* Total * 0.00000 1.99903 6.00000 0.00097 8.00000
(Occupancy) Bond orbital/ Coefficients/ Hybrids
--------------------------------------------------------------------------------
1. (1.99853) BD ( 1) B 1 - H 2
( 44.48%) 0.6669* B 1 s( 33.33%)p 2.00( 66.67%)
0.0000 0.5774 0.0000 0.0000 0.0000
0.8165 0.0000 0.0000 0.0000
( 55.52%) 0.7451* H 2 s(100.00%)
1.0000 0.0000
2. (1.99853) BD ( 1) B 1 - H 3
( 44.48%) 0.6669* B 1 s( 33.33%)p 2.00( 66.67%)
0.0000 0.5774 0.0000 0.7071 0.0000
-0.4082 0.0000 0.0000 0.0000
( 55.52%) 0.7451* H 3 s(100.00%)
1.0000 0.0000
3. (1.99853) BD ( 1) B 1 - H 4
( 44.48%) 0.6669* B 1 s( 33.33%)p 2.00( 66.67%)
0.0000 0.5774 0.0000 -0.7071 0.0000
-0.4082 0.0000 0.0000 0.0000
( 55.52%) 0.7451* H 4 s(100.00%)
1.0000 0.0000
4. (1.99903) CR ( 1) B 1 s(100.00%)
1.0000 0.0000 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.0000
5. (0.00000) LP*( 1) B 1 s(100.00%)
6. (0.00000) RY*( 1) B 1 s( 0.00%)p 1.00(100.00%)
Once opened in GaussView 5.0 an optionwas selected to colour the molecule according to electron density. Green indicates a relatively positive charge, whereas Red indicates a negative one. This can be seen in the diagram below, the Green colour of the central Boron and its positive value of 0.33 is in agreement with its well known Lewis Acidic behavior.

It can be seen from both the figure above and the natural population analysis that the electron density is equal in each of the Hydrogen atoms, which is to be expected since the molecule is highly symmetric.
Using the Occupancy data above it can also be seen from the LOG file that 44% of the electron density is deemed to be on the Boron and 56% on the Hydrogen of each B-H bond. In addition the s and p character of each bond can be seen to be 33%:67% (s:p), this is inidicative of sp2 hybridised bonds and is in agreement with the 120 degree bond angles found in the optimisation.
There is one error in the computational calculations above: while the 4th orbital makes sense the 5th orbital does not. The 4th orbital is a Boron 1s orbital so it should be expected to have 100% s character. However the data suggests the 5th orbital also has 100% s character whereas it should actually be an unoccupied p orbital, which is orbital 6 instead.
TlBr3
Geometry Optimisation of TlBr3
As in the BH3 optimisation the TlBr3 molecule was drawn with a trigonal planar geometry on GaussView 5.0. However on this occasion the LANL2DZ basis set was used instead of 321-G. The method was the same (DFT/ BLYP). The reason for this is that this basis set uses psuedo potentials which is necessary for larger metal atoms such as Thallium. The pseudo potential carries out the calculation solely on the valence electrons of the Thallium, which is an approximation but should be an accurate one for such a large atom. The LANL2DZ basis set is also larger than the 321-G basis set so should provide a more reliable result for a second row element such as Thallium.
The more relevant information from the LOG FILE is shown below, but it has also been published in full detail here.
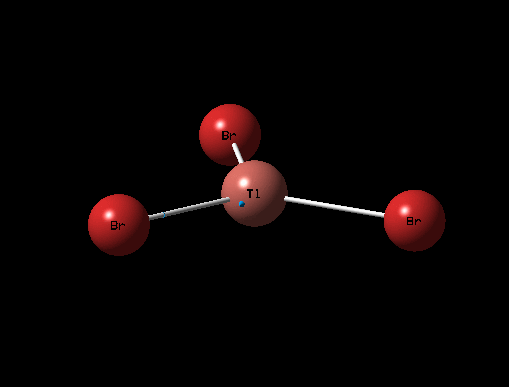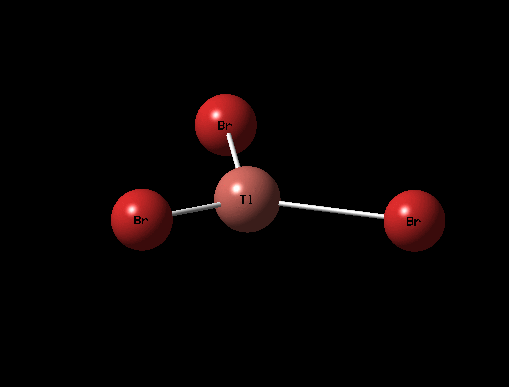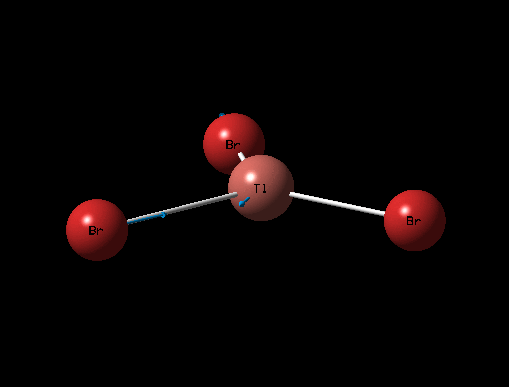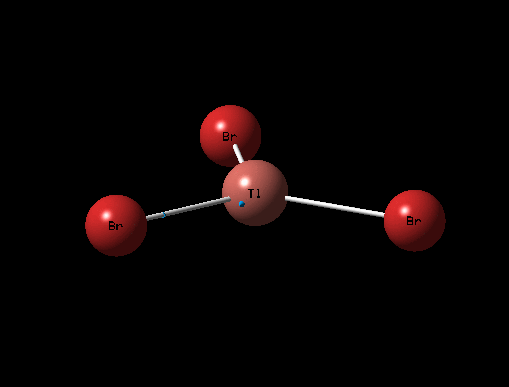
The 3D view of the optimised TlBr3 molecule can be seen here

Item Value Threshold Converged?
Maximum Force 0.000002 0.000450 YES
RMS Force 0.000001 0.000300 YES
Maximum Displacement 0.000022 0.001800 YES
RMS Displacement 0.000014 0.001200 YES
Predicted change in Energy=-6.083881D-11
Optimization completed.
-- Stationary point found.
Optimized Parameters
(Angstroms and Degrees)
-------------------- ----------------------
Name Definition Value Derivative Info.
------------------------------------------------------------------
R1 R(1,2) 2.651 -DE/DX = 0.0
R2 R(1,3) 2.651 -DE/DX = 0.0
R3 R(1,4) 2.651 -DE/DX = 0.0
A1 A(2,1,3) 120.0 -DE/DX = 0.0
A2 A(2,1,4) 120.0 -DE/DX = 0.0
A3 A(3,1,4) 120.0 -DE/DX = 0.0
D1 D(2,1,4,3) 180.0 -DE/DX = 0.0
------------------------------------------------------------------


It can be seen from the above that the optimisation was carried out in 3 iterations, and a stationary point was found.


The optimised structure for TlBr3 has bond length 2.65Å and 120degree bond angle this is confirmed by the Jmol and the diagrams above. Once again this suggests the molecule is sp2 hybridised.
Frequency Analysis

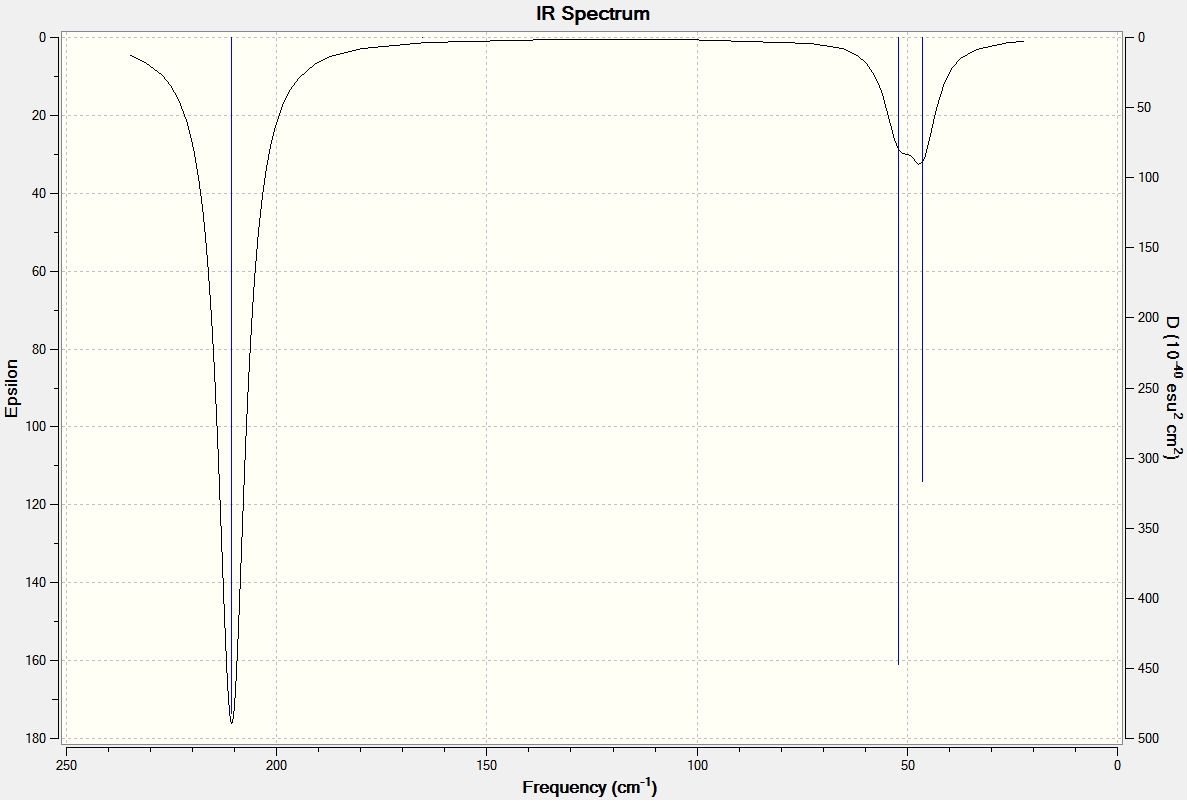
Once again the absence of negative absorptions confirms the geometry optimisation was successful in finding a minimum point.
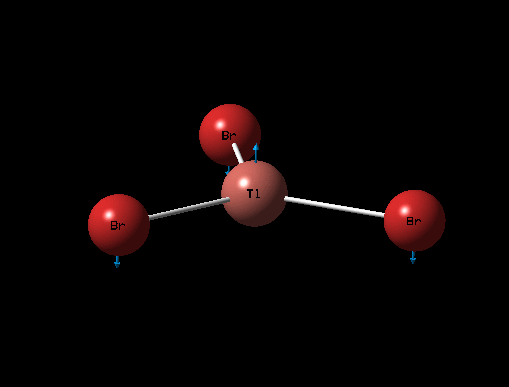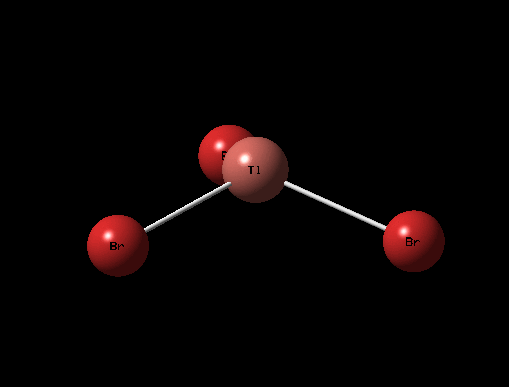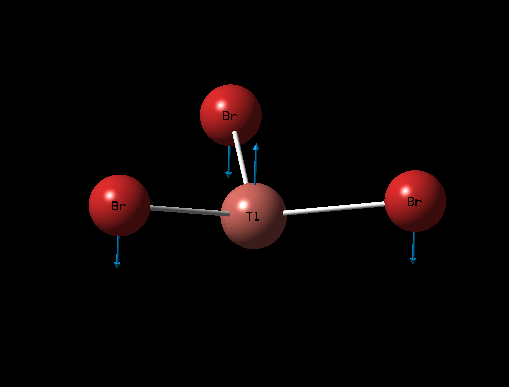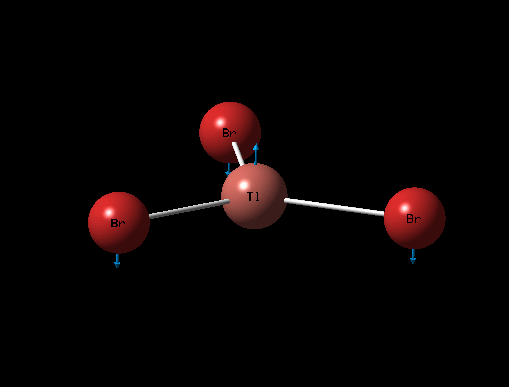
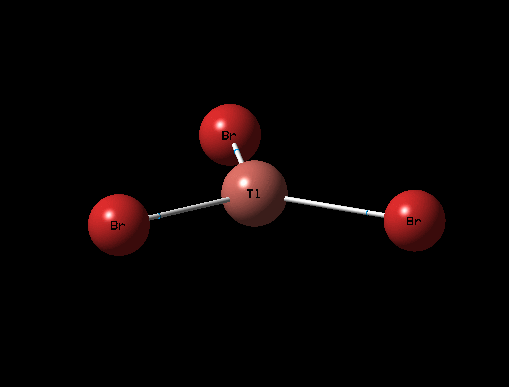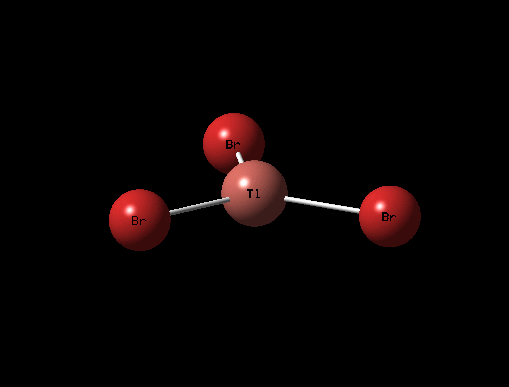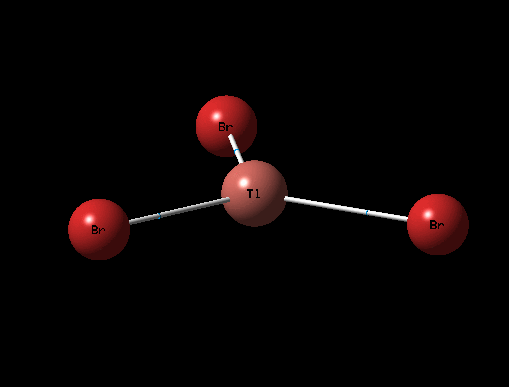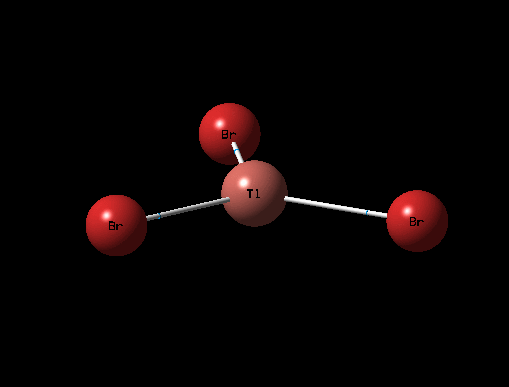
| Description for Mode of Vibration | Type of Vibrational Mode | Intensity of Absorption | Frequency of Absorption (cm-1) | Diagram | Link to 3D animation |
|---|---|---|---|---|---|
| Symmetric Tl-Br Scissoring | A2 " | 4 | 46 |  |
Show Animation |
| Asymmetric Tl-Br Rock | E' | 4 | 46 |  |
Show Animation |
| Umbrella Deformation | E' | 6 | 52 |  |
Show Animation |
| Symmetric Tl-Br Stretch | A1' | 0 | 165 |  |
Show Animation |
| Asymmetric B-H Stretch (1) | E' | 25 | 211 |  |
Show Animation |
| Asymmetric B-H Stretch (2) | E' | 25 | 211 |  |
Show Animation |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
It can be seen from the table above that there are 6 vibrational modes for TlBr3, and in the IR spectrum there are 2 main peaks, the lower frequency peak (4-6cm-1) is broad because 2 peaks have overlapped slightly, the shoulder at 4cm-1 corresponds to the A2" and E' (Asymmetric Tl-Br Rock) stretches which are degenerate. The two E' (B-H stretches) are completely degenerate and therefore are completely overlapped on spectrum and appear as one peak. The A' (Tl-Br) stretch is completely symmetric and so has an intensity of 0 and does not appear on the spectrum.
The data above shows good general agreement with the literature from the wet lab [5]. However this data can only be used as a rough guide since the spectrum was taken in water. Unfortunately no prior computational literature could be found for TlBr3 IR absorptions.
Analysis of the low frequencies shows that relative to the real absorptions they are small and close to zero suggesting the results are accurate. as discussed previously the closer the low frequency absorptions are to zero the more reliable the results.
Full mass-weighted force constant matrix: Low frequencies --- -3.4213 -0.0026 -0.0004 0.0015 3.9367 3.9367 Low frequencies --- 46.4289 46.4292 52.1449
What is a bond?
It can be seen from some of the Jmol diagrams and pictures of the molecules throughout this module that sometimes a 'bond' is not drawn between two atoms where you would expect one. This is simply because GuassView program has set parameters for when a bond is drawn or not based on how far atoms are apart. The majority of these parameters are based on organic molecules and bond lengths, since inorganic bonds tend to be longer in general there is a high chance GaussView will not draw a bond between many of the atoms. This prompts the question what really is a bond? after-all it is clearly not a line drawn between two atoms. In reality a bond is the attraction of 2 atoms, for covalent bonds this is through the overlap of their atomic orbitals in order that they can share electron density. In metallic bonds this is the delocalisation of free negatively charged electrons across positively charged nuclei. Clearly in reality bonding is far more complex than simply being bonded, double bonded or not bonded at all, as the attraction between atoms does not occur in these digital steps, rather the extent of bonding is on a continuous scale.
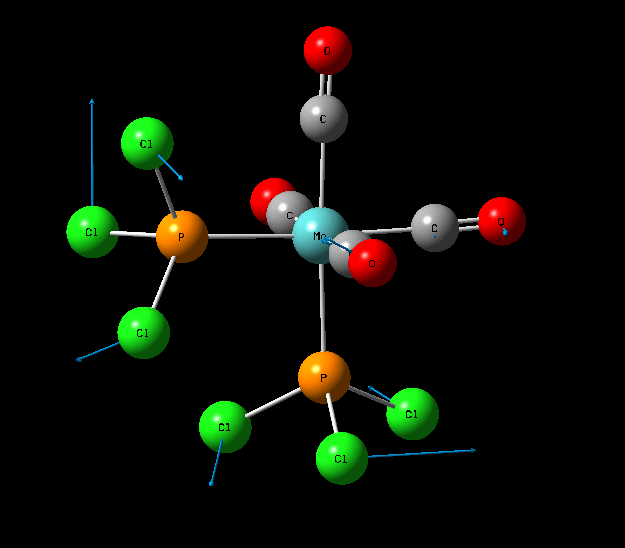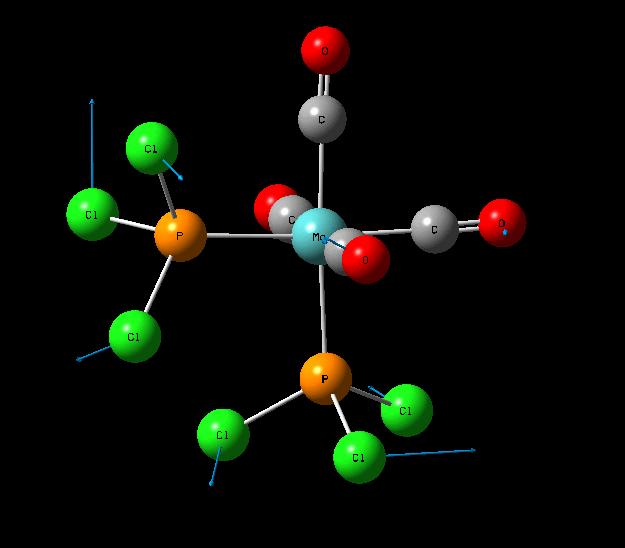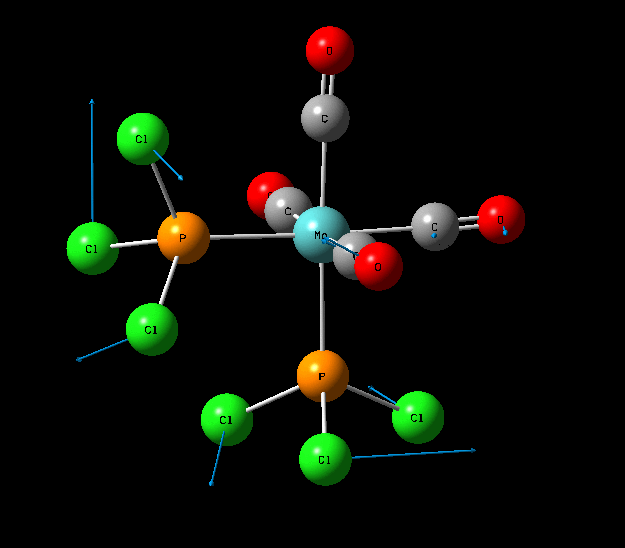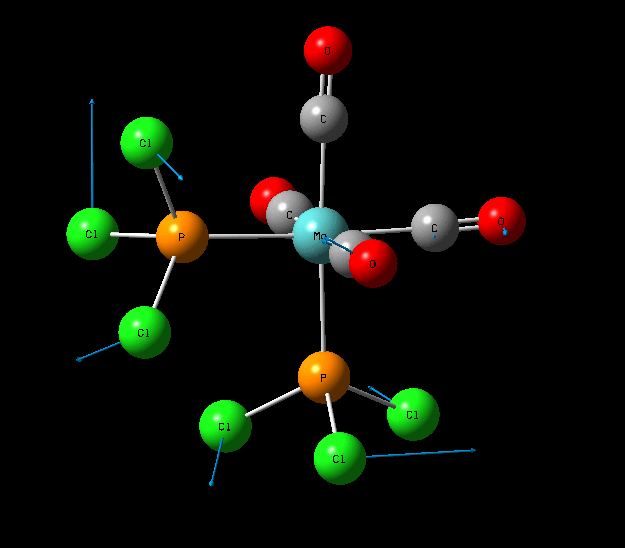
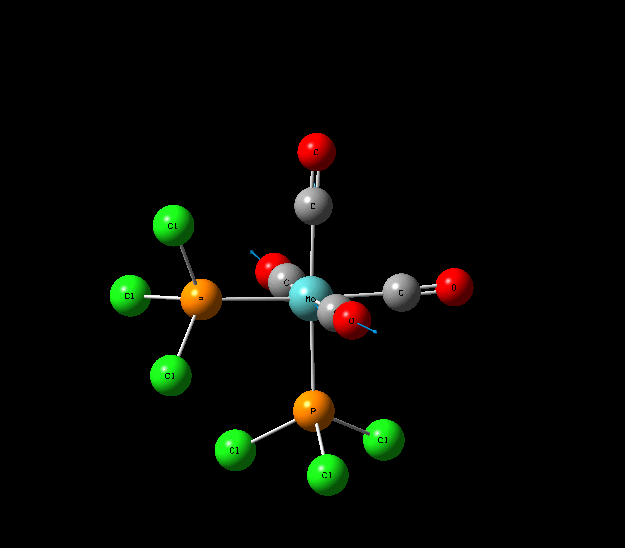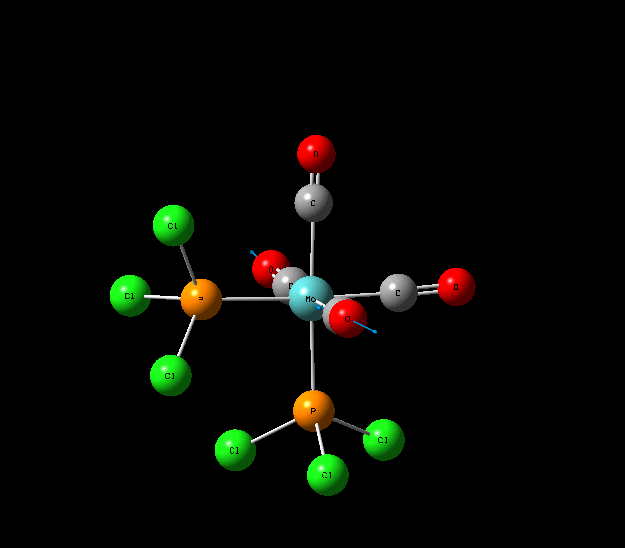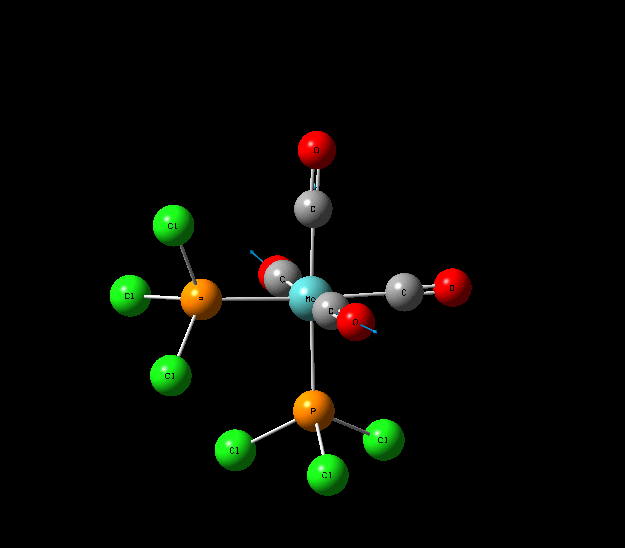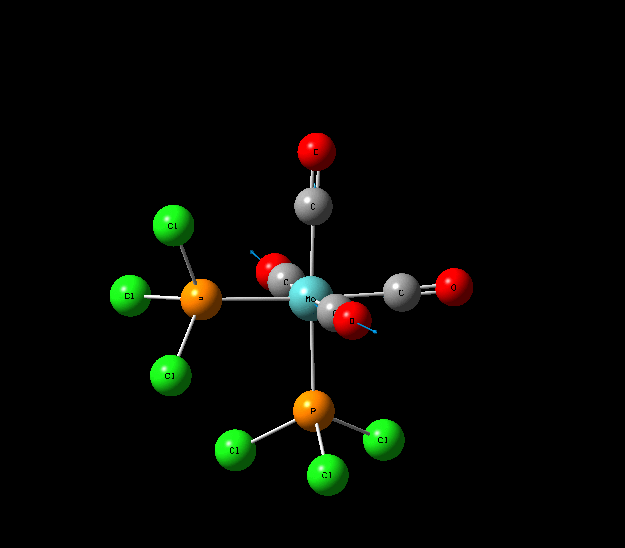
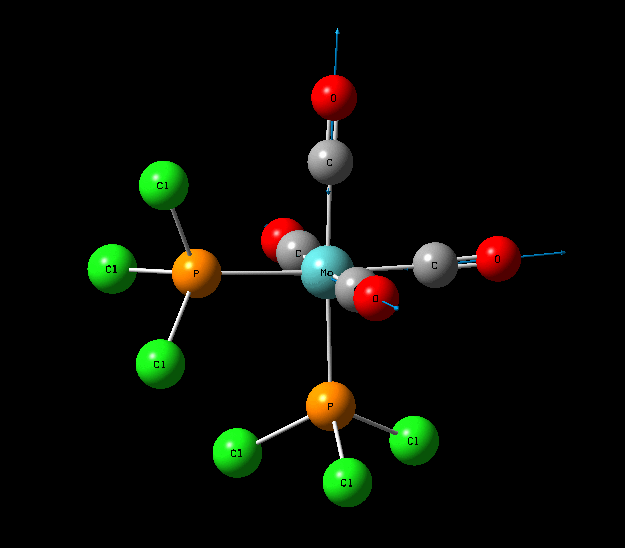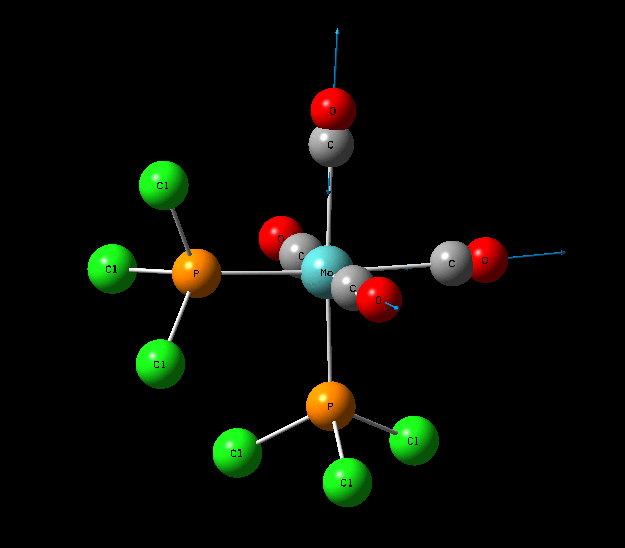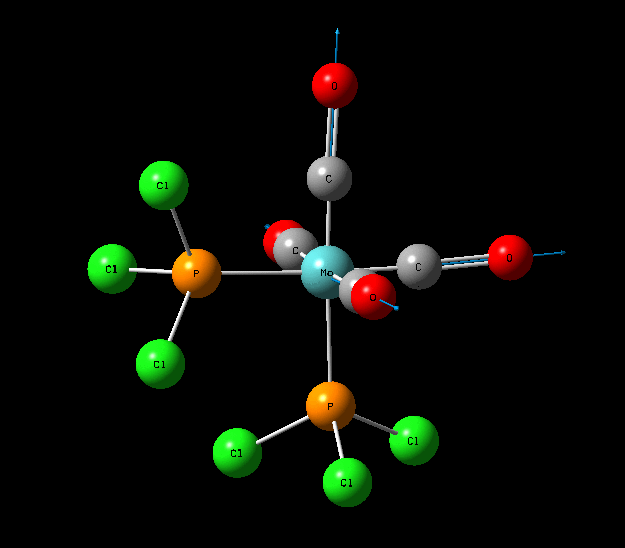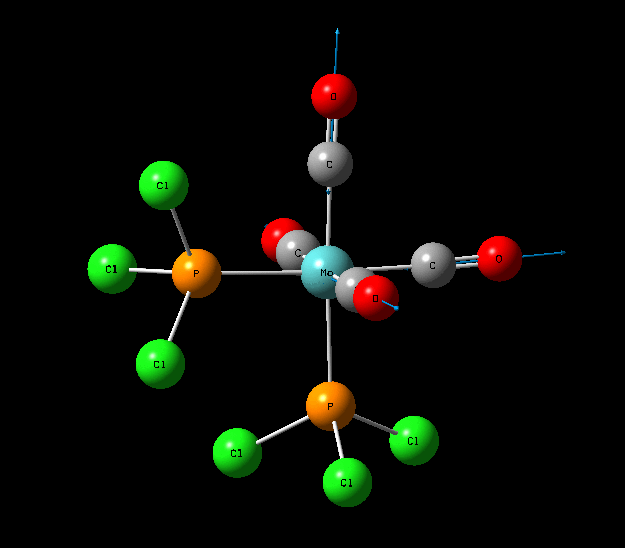
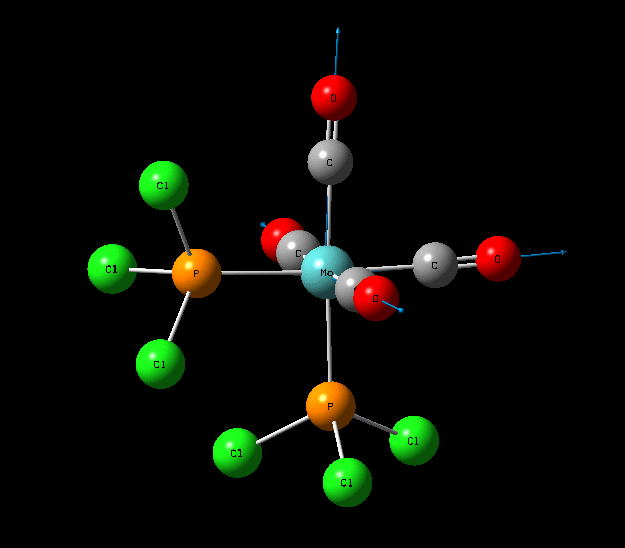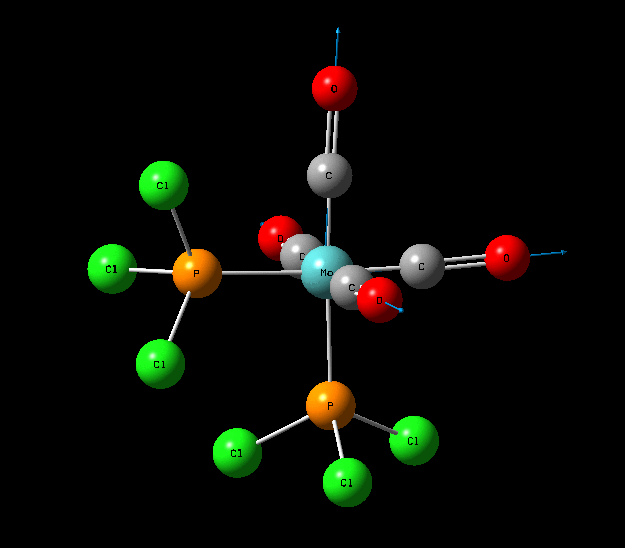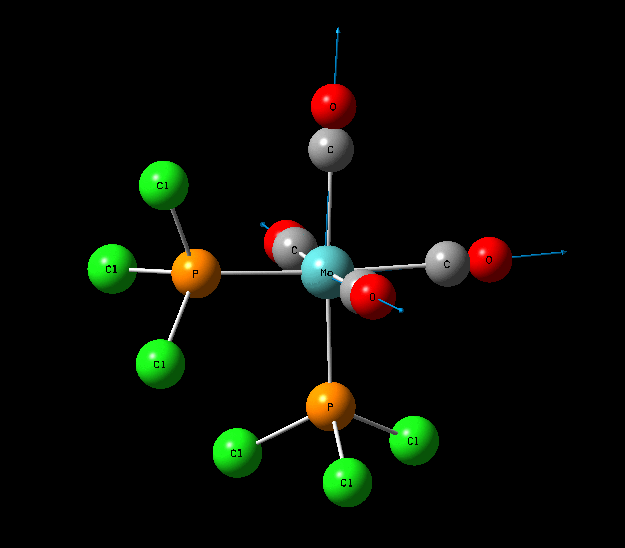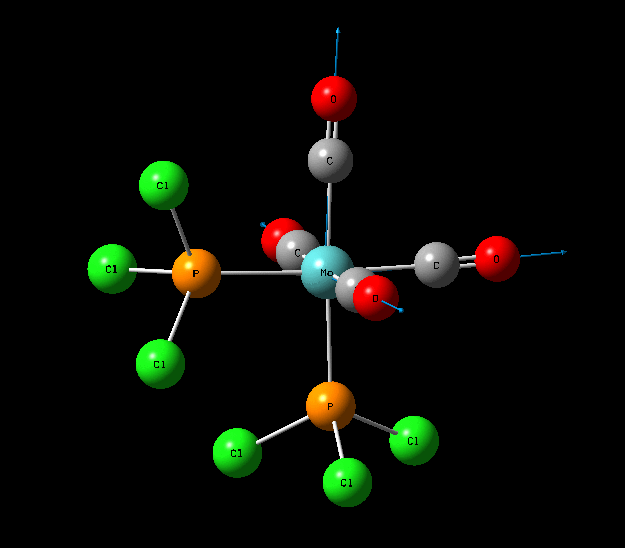
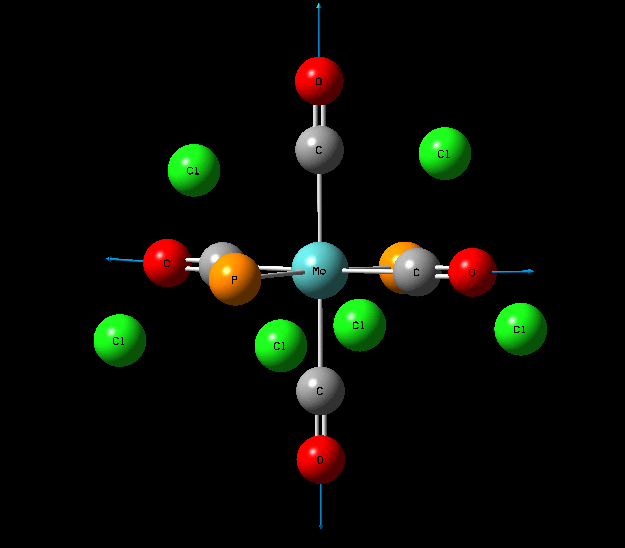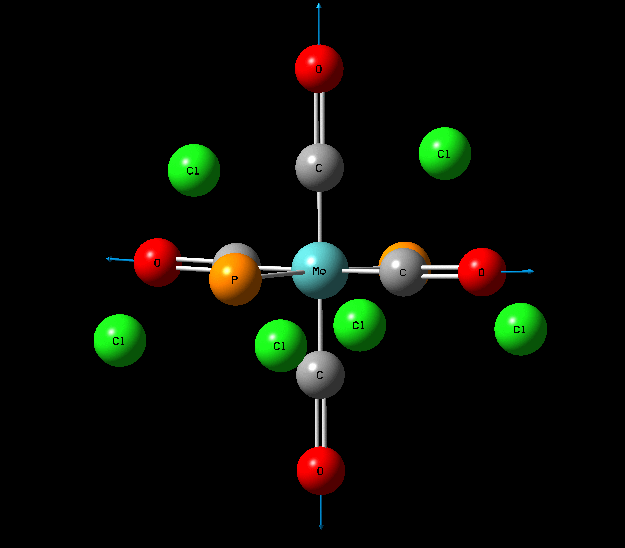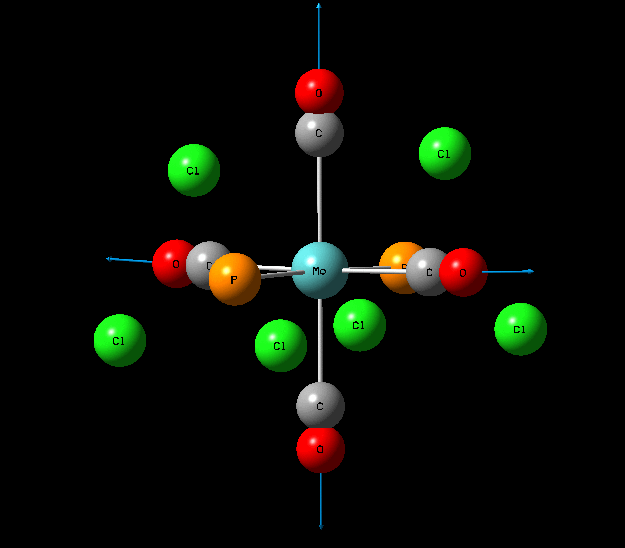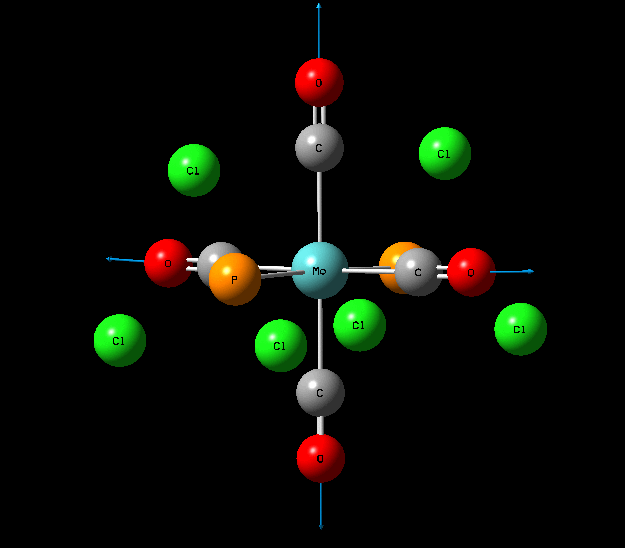
Isomers of Mo(CO)4L2
The optimisation of the both the cis, and trans isomers were carried by moving up the basis sets from LANL2MB to LANL2DZ to LANL2DZ + extrabasis set. Vibrational analysis will then be run on the fully optimised structure. During second year inorganic synthesis labs this same complex was studied in the wet lab, in that case the L ligand that was used was the more common PPh3. In an ideal scenario this same ligand would be used in the computational calculations, however the large number of electrons in the Phenyl rings would have a large computational cost therefore they are replaced with Cl ligands which is a good choice since it will have a similar steric demand and contribute to bonding in similar manner.
Geometry Optimisation of Mo(CO)4L2
LanL2MB Basis Set
Having drawn each of the structures in GaussView 5.0, a loose optimisation was run over the structures using the LanL2MB basis set, which was very quick because it a small basis set, the LOG file can be found here [6]. This is shown below.
cis - Mo(CO)4L2



Item Value Threshold Converged?
Maximum Force 0.000714 0.002500 YES
RMS Force 0.000192 0.001667 YES
Maximum Displacement 0.009868 0.010000 YES
RMS Displacement 0.003501 0.006667 YES
Predicted change in Energy=-4.775072D-06
Optimization completed.
-- Stationary point found.
trans - Mo(CO)4L2
LOG file [7]
Item Value Threshold Converged?
Maximum Force 0.000185 0.002500 YES
RMS Force 0.000056 0.001667 YES
Maximum Displacement 0.002972 0.010000 YES
RMS Displacement 0.001019 0.006667 YES
Predicted change in Energy=-6.046569D-07
Optimization completed.
-- Stationary point found.
LanL2DZ Basis Set
cis LOG file [8], trans LOG file [9].
The loosely optimised structures were then manually altered using the 'Dihedral Angle' function on GaussView 5.0. This was done so that future optimisation would not find a false minimum, rather than the global minimum. The trans isomer was made to have eclipsed PCl3 groups by making the dihedral angle between Chlorine atoms 0 degrees. For the cis isomer the dihedral angle between one Cl atom and a carbonyl was forced to be 180degrees and then this same carbonyl group was given a 0 degree dihedral angle with a Cl atom on the other PCl3 group.
Having completed this manual alteration both structures were re-optimised with the higher LanL2DZ basis set, the calculations were given the additional words 'int=unltrafine scf=conver=9' in the Gaussian Calculation Set-up. This is shown below.
cis - Mo(CO)4L2



Item Value Threshold Converged?
Maximum Force 0.000016 0.000450 YES
RMS Force 0.000005 0.000300 YES
Maximum Displacement 0.000989 0.001800 YES
RMS Displacement 0.000270 0.001200 YES
Predicted change in Energy=-7.775720D-09
Optimization completed.
-- Stationary point found.
trans - Mo(CO)4L2
Item Value Threshold Converged?
Maximum Force 0.000037 0.000450 YES
RMS Force 0.000012 0.000300 YES
Maximum Displacement 0.000976 0.001800 YES
RMS Displacement 0.000169 0.001200 YES
Predicted change in Energy=-3.384020D-08
Optimization completed.
-- Stationary point found.
LanL2DZ plus Extra Basis Set, Including contribution of Phosphorus d Atomic Orbitals
cis LOG file [10], trans LOG file [11].
cis - Mo(CO)4L2



Item Value Threshold Converged?
Maximum Force 0.000138 0.000450 YES
RMS Force 0.000044 0.000300 YES
Maximum Displacement 0.001079 0.001800 YES
RMS Displacement 0.000409 0.001200 YES
Predicted change in Energy=-2.380857D-07
Optimization completed.
-- Stationary point found.
trans - Mo(CO)4L2
Item Value Threshold Converged?
Maximum Force 0.000027 0.000450 YES
RMS Force 0.000006 0.000300 YES
Maximum Displacement 0.000336 0.001800 YES
RMS Displacement 0.000081 0.001200 YES
Predicted change in Energy=-9.511944D-09
Optimization completed.
-- Stationary point found.
Structural Analysis of Optimized Geometries
| Bond | Cis Isomer Bond Length (A) | Trans Isomer Bond Length (A) | Lit. Bond Length (cis) [12] | Percentage Error (%)(cis) | Lit. Bond Length (trans)[13] | Percentage Error (trans) (%) |
|---|---|---|---|---|---|---|
| C=O | 1.17, 1.18 | 1.17, 1.18 | 1.15 | 2 | 1.15 | 2 |
| P-Cl | 2.12 | 2.12 | - | - | - | - |
| Mo-C | 2.02, 2.05 | 2.06 | 1.98, 2.05 | 0 | 1.87 | 10 |
| Mo-P | 2.48 | 2.42 | 2.56 | 3 | 2.37 | 2 |
N.B The literature values for the cis isomer correspond to a Mo(CO)4(PPhMe2)(PPh3) molecule, whereas the literature values for the trans isomer correspond to a Mo(CO)4(PPh3)2 molecule. This small difference in ligands should have little effect on the bond lengths seen above.
It can be seen from the percentage differences above that the results are in good agreement with the literature. A look at the bond lengths in the Jmol diagram reveals that as the optimisation is carried at higher level basis set the bond lengths in general shorten and become closer to the literature values seen. It can be concluded from this that taking into account the low lying d orbitals of heavier elements is worthwhile.
Analysis of Stabilities of the Isomers
From the summary files of the optimisation of each of the isomers the relative energies of the molecules can be used to predict which isomer is more stable, and therefore the thermodynamic product. It can be seen that after the final optimisation (including the extra basis set) the trans isomer is more stable, -623.6942 Hartrees, compared with -623.6929 Hartrees for the cis isomer. A conversion to the more familiar KJ/mol units can be carried by multiplying by 2625.50 [14]. This gives an energy difference of 3.41KJ/mol in favour of the trans isomer. This result is consistent with literature and the second year synthesis labs which found that the cis isomer is converted to the trans isomer at elevated temperatures. However the difference found computationally is not satisfactory to come to this conclusion since it is known that there is roughly a 10KJ/mol error in these calculations [15].
The results do though show that taking account of the phosphorus d orbitals using the extrabasis made a big difference to the results: without the extra basis set optimisation it would have been found that the cis isomer is the most stable form by computational calculations.
Despite the possible error in the calculations, the final result is that the trans-isomer is more stable and this intuitively makes sense due to the high steric demand of the PPh3/ PCl3 groups. Further increasing the size of these groups would accentuate the difference in energy. The only way the cis isomer could become the thermodynamic product would be to introduce a stabilising interaction between the 2 phosphorus groups, this could be achieved by adding an amine or hydroxyl group on one of the groups to enable Hydrogen bonding.
Frequency Analysis
cis LOG file [16], trans LOG file [17].
The fully optimised structures for both isomers were sent back to the HPC servers to find their vibrational frequencies. The absence of negative absorptions in both spectra (the lowest frequency absorptions are shown), indicate the optimisations carried out previously were successful.
cis - Mo(CO)4L2

| Mode of Vibration | Type of Vibrational Mode | Intensity of Absorption | Frequency of Absorption (cm-1) | Literature value for Frequency (cm-1)[18] | Percentage Error (%) | Diagram | Link to 3D animation |
|---|---|---|---|---|---|---|---|
| Rotation of the PCl3 groups | - | 0 | 12 | - | - |  |
Show Animation |
| Rotation of the PCl3 groups | - | 0 | 20 | - | - |  |
Show Animation |
| Asymmetric Carbonyl Stretch (1) | B2' | 1605 | 1938 | 1874 | 3 |  |
Show Animation |
| Asymmetric Carbonyl Stretch (2) | B2 | 813 | 1942 | 1910 | 2 |  |
Show Animation |
| Stretch for all 4 Carbonyls (asymmetric) | A1' | 588 | 1952 | 1926 | 1 |  |
Show Animation |
| Stretch for all 4 Carbonyls (symmetric) | A1' | 544 | 2019 | 2021 | 0 |  |
Show Animation |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
It can be seen from the above data that the cis isomer has 4 unique absorption corresponding to C=O stretches. The B2' and B2 stretches are overalaid on the IR spectrum and so can not be resolved from each other. This can be seen clearly in zoomed in IR spectrum below. The computational calculations agree well literature from the wet lab with the largest percentage difference seen above is 3%.


trans - Mo(CO)4L2
| Description for Mode of Vibration | Type of Vibrational Mode | Intensity of Absorption | Frequency of Absorption (cm-1) | Literature value for Frequency (cm-1)[19] | Percentage Error (%) | Diagram | Link to 3D animation |
|---|---|---|---|---|---|---|---|
| Symmetric Rotation of PCl3 bonds | - | 5 | 46 | - | - | Show Animation | |
| Asymmetric Rotation of PCl3 bonds | - | 6 | 46 | - | - | Show Animation | |
| Asymmetric trans -Carbonyl stretch (1) | Eu | 1475 | 1950 | 1887 | 3 | Show Animation | |
| Asymmetric trans -Carbonyl stretch (2) | Eu | 1467 | 1951 | 1887 | 3 | Show Animation | |
| Stretching Mode for all Carbonyls | B1g | 1 | 1977 | - | - | Show Animation | |
| Stretching Mode for all Carbonyls | A1g | 4 | 2031 | - | - | Show Animation |
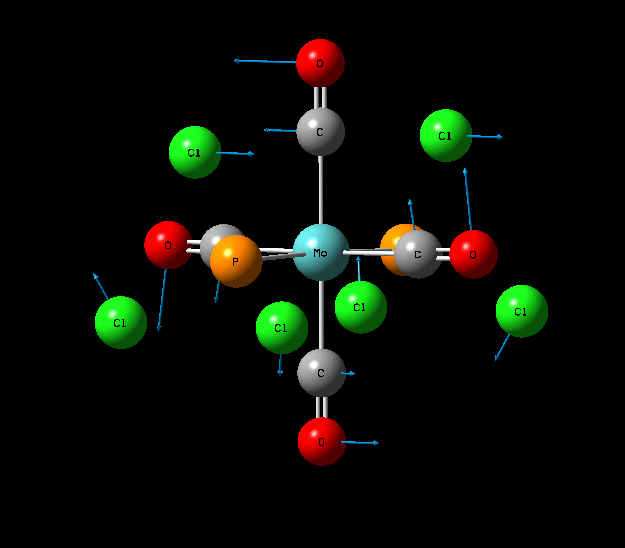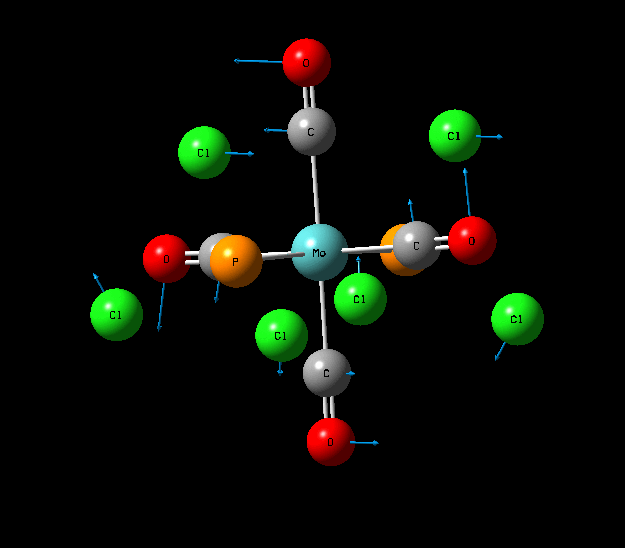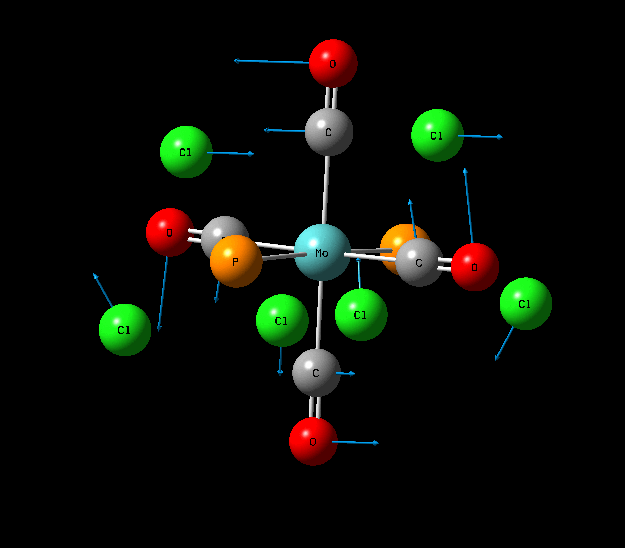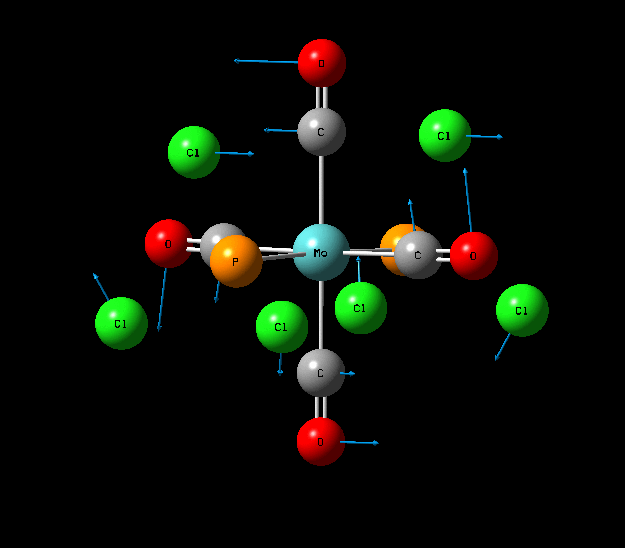{kind=link}
{kind=link}
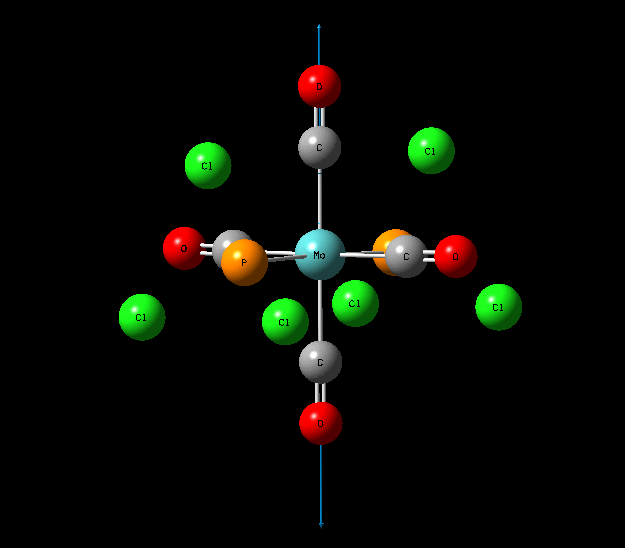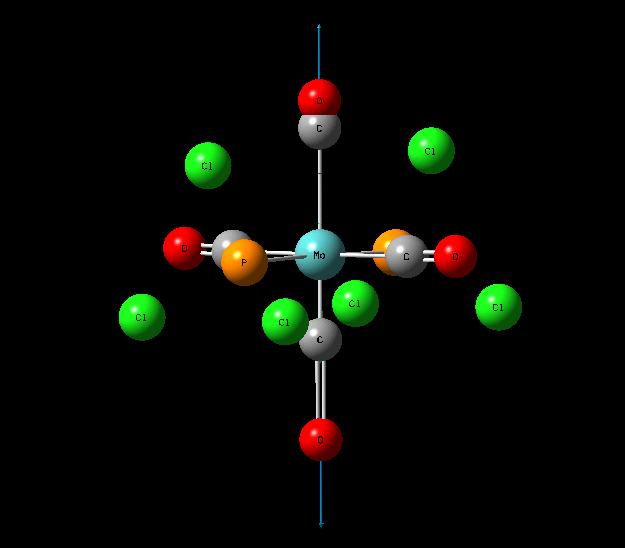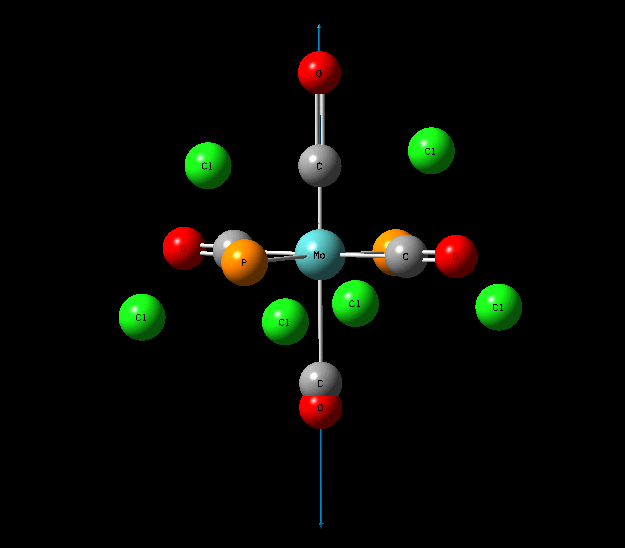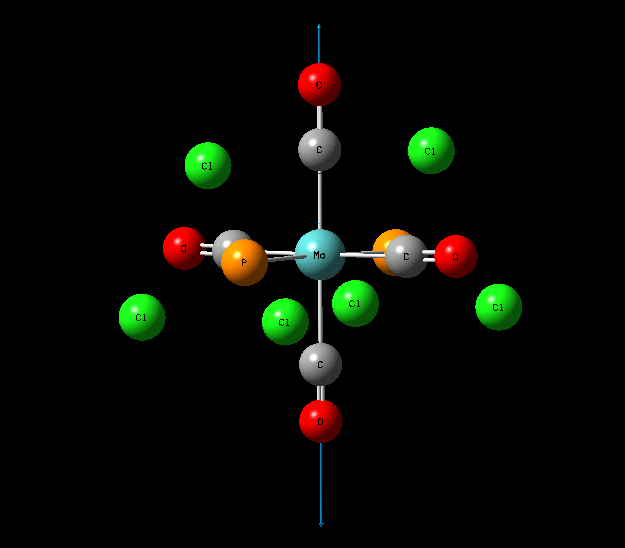{kind=link}
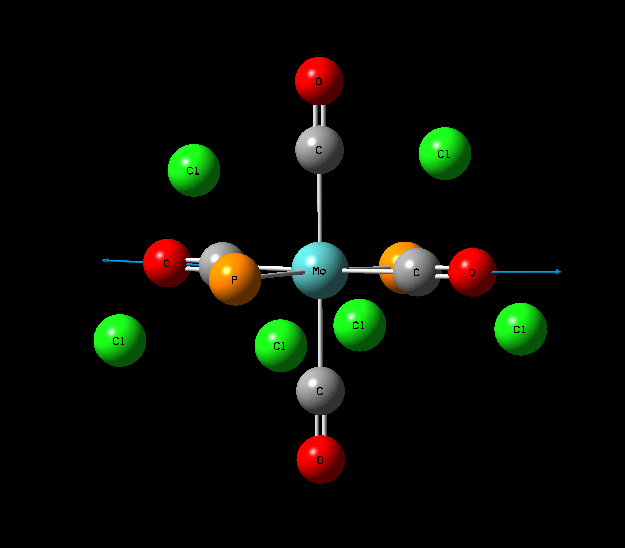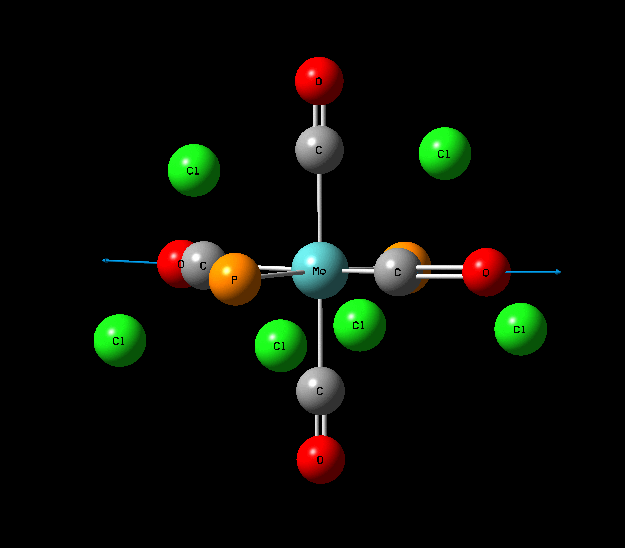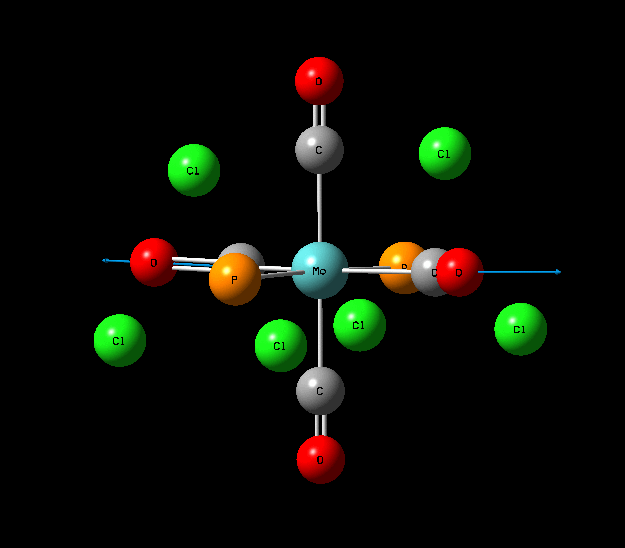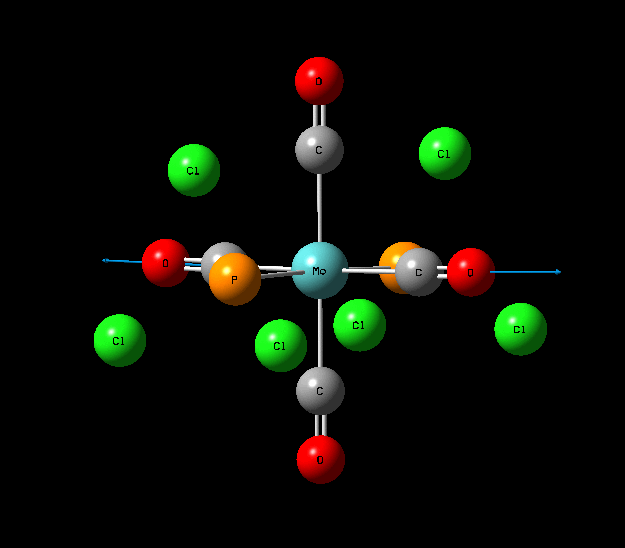{kind=link}
{kind=link}
{kind=link}
In contrast with the cis isomer, the trans isomer only has 1 unique vibrational absorption at 1950cm-1. This peak corresponds to the two E stretches which are degenerate. The absorptions at 1977cm-1 and 2031cm-1 are not seen in the vibrational spectrum because they are highly symmetric, little to no dipole moment being formed and so a very low intensity. The single peak corresponding to the C=O functionality can be seen clearly in the zoomed in IR spectrum. Again the computational calculations agree well literature from the wet lab with a low percentage difference.
Mini Project - Aromaticity in Metal Clusters
Introduction
In recent years it was noticed by Li et al that all metal clusters such as Al42- can exhibit a large amount of resonance in a planar 4-membered ring, and that this aromaticity is not the same as that seen in normal Huckel aromatics such as C4H42+. By doing a count of each of the species valence electrons it is clear that they are not aromatic: the C4H42+ 18 electrons whereas the all metal M42- species have only 14 electrons. The carbon di-cation exhibits classical bonding with 8 2-centre, 2-electron bonds (4 C-C + 4 C-H) and one 4-centre π aromatic bond, giving the molecule 4n+2 Huckel aromaticity. The metal clusters on the other hand are fundamentally electron deficient which gives rise to their unusual bonding: 8 electrons are taken up due to each metal having a lone pair, 2 are used in a 4-centre 2-electron π bond giving the metal cluster its π aromaticity. This leaves 4 electrons for 4 σ which is clearly not possible in a classical sense. It was found by Fowler et al. [20] that in Al42- it is the σ electrons in the ring that are responsible for a diamagnetic current induced by an external magnetic field. This is strong evidence to suggest that species such as Al42- are both σ and π aromatic. In this Mini Project several isomers of Na2Al4 will be investigated using the computational techniques learnt during the previous parts of the module. The role of the sodium is simply to stabilize the otherwise unstable aluminium dianion, this should not effect the electronic structure of the M42- dianion to a large extent.[21]
The aim is to first to optimise the isomers of Aluminium and Gallium to find the most thermodynamically favoured arrangement, and once these are found to use MO and NBO analyses to illustrate the presence of both π and σ aromaticity.
Optimisations
First the structures were given a loose optimisation using the LanL2DZ pseudo potential basis set. Following this the 6113G(d) basis set was used to optimise the structures taking into account all the electrons and the d orbitals. This optimisation was completed an all the isomers, from these the D4h (square), Cs and D4h (pyramidal) were selected to be further optimised using the MP2 method.
The summary and LOG files for each of the optimisations is shown below:
| D4h (square) | Cs | D4h (pyramidal) | C2v | D2h | |
|---|---|---|---|---|---|
| LanL2DZ | - |  |
 |
 |
 |
| BLYP/ 6113-G(d) |  |
 |
 |
 |
 |
| MP2/ 6113-G(d) |  |
 |
 |
- | - |
| D4h (square) | Cs | D4h (pyramidal) | C2v | D2h | |
|---|---|---|---|---|---|
| LanL2DZ | - |  |
 |
 |
 |
| BLYP/ 6113-G(d) |  |
 |
 |
 |
 |
| MP2/ 6113-G(d) |  |
 |
 |
- | - |
It can seen from the energies in the summary files and the Jmol diagrams that in general as you move down the table the energy of the molecules goes down, and the bond lengths in the structures goes down.
It can also be seen that the energy of the Cs isomer is the most thermodynamically stable in the case of both aluminium and gallium. The energies produced by the calculations as a whole are in perfect agreement with the literature, with the D4h (pyramidal) structure being the second most stable isomer. For both Gallium and Aluminium the D2h isomer was clearly the least stable isomer by some margin.
It should be noted that the Gallium isomers were all more stable than there Aluminium counterparts due to their more diffuse atomic orbitals allowing for greater overlap.
Since the Cs and D4h (pyramidal) isomers are the most stable these geometries are concentrated on most for the future calculations.
Molecular Orbital Analysis
| D4h (square) [46] | Cs [47] | D4h (pyramidal) [48] | C2v [49] | D2h [50] | |
|---|---|---|---|---|---|
| LUMO |  |
 |
 |
 |
 |
| HOMO |  |
 |
 |
 |
 |
| HOMO-1 |  |
 |
 |
 |
 |
| HOMO-2 |  |
 |
 |
 |
 |
| HOMO-3 |  |
 |
 |
 |
 |
| HOMO-4 |  |
 |
 |
 |
 |
| HOMO-5 |  |
 |
 |
 |
 |
| HOMO-6 | - |  |
 |
 |
 |
| D4h (square)[51] | Cs [52] | D4h (pyramidal) [53] | C2v [54] | D2h[55] | |
|---|---|---|---|---|---|
| LUMO |  |
 |
 |
 |
 |
| HOMO |  |
 |
 |
 |
 |
| HOMO-1 |  |
 |
 |
 |
 |
| HOMO-2 |  |
 |
 |
 |
 |
| HOMO-3 |  |
 |
 |
 |
 |
| HOMO-4 |  |
 |
 |
 |
 |
| HOMO-5 |  |
 |
 |
 |
 |
| HOMO-6 | - |  |
 |
 |
 |
The MO diagrams above correlate well with those seen in the literature.[56] It can be seen that in general the MO's have a very similar structure for all of the isomers, although the order of them is different between some of the isomers. This is an indication that the sodium does indeed have little effect on the electronic structure of the dianion. In addition the MOs are clearly very similar for the Aluminium and Gallium which is to be expected.
It can be seen from the MO diagrams that the HOMO corresponds to the π aromatic electrons across the 4 metal centres. The HOMO-3 to HOMO-6 molecular orbitals correspond to the Aluminium / Gallium lone pairs. Finally the HOMO-1 and HOMO-2 molecular orbitals correspond to the σ bonding orbitals.
A comparison between the MOs above and the MOs for C4H42+ shows that the electronic structures of these molecules is quite different. Most importantly there is a difference between the HOMO-2 of each of the species, the carbon ring has a HOMO-2 which corresponds to π bond, whereas the HOMO-2 in the metal clusters corresponds to an aromatic σ bonding interaction. This will be analysed further in the NBO analyses.
| HOMO | HOMO-1 | HOMO-2 | HOMO-3 | HOMO-4 | HOMO-5 | HOMO-6 |
|---|---|---|---|---|---|---|
 |
 |
 |
 |
 |
 |
 |
Effect of Sodium
To analyse the stabilising effect of sodium on the dianion the energy of a lone sodium atom was found by optimising its structure GaussView 5.0. This was found to be -161.8460 a.u. Next the energy of the Al42- fragment is taken from the earlier optimisation, =-7699.4640. The energy of these fragments added together is then compared with the energy of a molecule which is bonded to the sodium atoms (-1291.6439), to reveal the stability gained from the sodium.
Therefore Stabilisation Energy = -1291.6439 - ((2 x -161.8460)-967.7997)= -0.1522 a.u. or -399KJ/mol.
Clearly this is a very stabilising interaction.
NBO Analyses
| D4h (square) | Cs | D4h (pyramidal) | |
|---|---|---|---|
| NBO |  |
 |
 |
| D4h (square) | Cs | D4h (pyramidal) | |
|---|---|---|---|
| NBO |  |
 |
 |
It can be seen from the NBOs above that the electrons are spread fairly evenly across the molecules, this is particularly evident in the D4h square molecules, where the electrons are spread exactly evenly, which is to be expected since the molecules are so symmetric. As a result of its higher electronegativity there is more electron density on the Boron atoms than the Sodium atoms in general
N.B some data has been removed from the Second Order Perturbation information shown below so that the more relevant and important data is clearer.
Analysis of Aluminium Cs
Second Order Perturbation Theory Analysis of Fock Matrix in NBO Basis
Threshold for printing: 0.50 kcal/mol
(Intermolecular threshold: 0.05 kcal/mol)
E(2) E(j)-E(i) F(i,j)
Donor NBO (i) Acceptor NBO (j) kcal/mol a.u. a.u.
===================================================================================================
within unit 1
1. BD ( 1)Al 1 -Al 2 /176. BD*( 1)Al 1 -Al 3 41.54 0.34 0.108
1. BD ( 1)Al 1 -Al 2 /179. BD*( 1)Al 2 -Al 4 41.54 0.34 0.108
2. BD ( 1)Al 1 -Al 3 /175. BD*( 1)Al 1 -Al 2 25.63 0.34 0.089
2. BD ( 1)Al 1 -Al 3 /177. BD*( 1)Al 1 -Al 4 26.99 0.36 0.093
2. BD ( 1)Al 1 -Al 3 /178. BD*( 1)Al 2 -Al 3 22.75 0.36 0.086
2. BD ( 1)Al 1 -Al 3 /180. BD*( 1)Al 3 -Al 4 48.56 0.30 0.112
3. BD ( 1)Al 1 -Al 4 /176. BD*( 1)Al 1 -Al 3 45.36 0.38 0.121
3. BD ( 1)Al 1 -Al 4 /179. BD*( 1)Al 2 -Al 4 21.18 0.38 0.083
4. BD ( 1)Al 2 -Al 3 /176. BD*( 1)Al 1 -Al 3 21.18 0.38 0.083
4. BD ( 1)Al 2 -Al 3 /179. BD*( 1)Al 2 -Al 4 45.36 0.38 0.121
5. BD ( 1)Al 2 -Al 4 /175. BD*( 1)Al 1 -Al 2 25.63 0.34 0.089
5. BD ( 1)Al 2 -Al 4 /177. BD*( 1)Al 1 -Al 4 22.75 0.36 0.086
5. BD ( 1)Al 2 -Al 4 /178. BD*( 1)Al 2 -Al 3 26.99 0.36 0.093
5. BD ( 1)Al 2 -Al 4 /180. BD*( 1)Al 3 -Al 4 48.56 0.30 0.112
6. BD ( 1)Al 3 -Al 4 /176. BD*( 1)Al 1 -Al 3 35.26 0.38 0.106
6. BD ( 1)Al 3 -Al 4 /179. BD*( 1)Al 2 -Al 4 35.26 0.38 0.106
39. LP ( 1)Al 3 / 37. LP*( 1)Al 1 19.88 0.02 0.030
39. LP ( 1)Al 3 / 38. LP*( 1)Al 2 154.65 0.02 0.083
40. LP*( 1)Al 4 / 37. LP*( 1)Al 1 154.65 0.02 0.083
40. LP*( 1)Al 4 / 38. LP*( 1)Al 2 19.88 0.02 0.030
37. LP*( 1)Al 1 / 41. LP*( 1)Na 5 46.05 0.03 0.056
38. LP*( 1)Al 2 / 41. LP*( 1)Na 5 46.05 0.03 0.056
39. LP ( 1)Al 3 / 41. LP*( 1)Na 5 39.90 0.06 0.064
40. LP*( 1)Al 4 / 41. LP*( 1)Na 5 39.90 0.06 0.064
6. BD ( 1)Al 3 -Al 4 / 45. LP*( 1)Na 6 22.78 0.58 0.105
45. LP*( 1)Na 6 /174. RY*( 21)Na 6
The data below is strong evidence for the resonance and σ aromaticity discussed previously. It can be seen from line 39. that there is a very large stabilizing interaction of 155kcal/mol corresponding to the donation of electrons from the lone pair on Al(3) into the antibonding lone pair on Al(2). An equal interaction is seen from the antibonding lone pair on Al(1) into the antibonding Al(2). It can be seen that in general there is a large amount of stabilisation gained in the molecule by resonance.
(Occupancy) Bond orbital/ Coefficients/ Hybrids
---------------------------------------------------------------------------------
1. (1.73592) BD ( 1)Al 1 -Al 2
( 50.00%) 0.7071*Al 1 s( 37.15%)p 1.68( 62.51%)d 0.01( 0.34%)
0.0000 -0.0011 0.6094 0.0033 -0.0080
0.0013 -0.0001 0.0003 -0.0154 -0.0039
0.0078 -0.0022 -0.0001 -0.0003 -0.3007
0.0143 -0.0135 -0.0001 0.0003 0.0000
-0.7307 0.0102 0.0005 -0.0059 0.0000
0.0006 0.0067 0.0059 0.0042 0.0574
( 50.00%) 0.7071*Al 2 s( 37.15%)p 1.68( 62.51%)d 0.01( 0.34%)
0.0000 -0.0011 0.6094 0.0033 -0.0080
0.0013 -0.0001 0.0003 -0.0154 -0.0039
0.0078 -0.0022 -0.0001 -0.0003 -0.3007
0.0143 -0.0135 -0.0001 0.0003 0.0000
0.7307 -0.0102 -0.0005 0.0059 0.0000
0.0006 -0.0067 -0.0059 0.0042 0.0574
2. (1.60474) BD ( 1)Al 1 -Al 3
( 39.74%) 0.6304*Al 1 s( 19.32%)p 4.16( 80.40%)d 0.01( 0.28%)
0.0000 -0.0009 0.4377 0.0330 0.0226
-0.0014 0.0000 -0.0002 0.0495 0.0140
-0.0029 -0.0006 -0.0002 -0.0012 -0.6380
-0.0544 0.0011 -0.0048 -0.0005 0.0005
0.6249 0.0276 0.0018 0.0007 0.0002
0.0093 -0.0094 0.0392 0.0298 -0.0156
( 60.26%) 0.7763*Al 3 s( 16.06%)p 5.21( 83.72%)d 0.01( 0.22%)
0.0000 -0.0001 0.3997 0.0284 0.0008
-0.0010 0.0002 -0.0002 0.0492 0.0068
0.0119 0.0034 -0.0002 -0.0002 0.6527
0.0284 0.0131 0.0018 0.0006 -0.0006
-0.6384 0.0048 0.0066 -0.0074 0.0000
-0.0060 0.0091 0.0350 0.0126 -0.0266
3. (1.75442) BD ( 1)Al 1 -Al 4
( 47.34%) 0.6880*Al 1 s( 43.69%)p 1.28( 55.97%)d 0.01( 0.35%)
0.0000 -0.0010 0.6599 -0.0360 -0.0107
0.0008 0.0000 0.0003 -0.0135 -0.0117
0.0078 -0.0020 -0.0001 0.0003 0.7011
-0.0071 -0.0032 0.0125 0.0002 0.0005
0.2591 -0.0184 0.0118 -0.0008 0.0000
-0.0065 0.0004 -0.0006 -0.0485 -0.0326
( 52.66%) 0.7257*Al 4 s( 42.42%)p 1.35( 57.30%)d 0.01( 0.28%)
0.0000 -0.0008 0.6510 -0.0075 0.0172
0.0006 -0.0001 0.0003 0.0116 -0.0096
-0.0049 -0.0007 -0.0001 -0.0001 -0.6938
-0.0184 0.0099 -0.0033 -0.0004 0.0000
0.3008 -0.0177 -0.0143 -0.0008 -0.0001
0.0032 -0.0017 -0.0108 -0.0441 -0.0268
4. (1.75442) BD ( 1)Al 2 -Al 3
( 47.34%) 0.6880*Al 2 s( 43.69%)p 1.28( 55.97%)d 0.01( 0.35%)
0.0000 -0.0010 0.6599 -0.0360 -0.0107
0.0008 0.0000 0.0003 -0.0135 -0.0117
0.0078 -0.0020 -0.0001 0.0003 0.7011
-0.0071 -0.0032 0.0125 0.0002 -0.0005
-0.2591 0.0184 -0.0118 0.0008 0.0000
-0.0065 -0.0004 0.0006 -0.0485 -0.0326
( 52.66%) 0.7257*Al 3 s( 42.42%)p 1.35( 57.30%)d 0.01( 0.28%)
0.0000 -0.0008 0.6510 -0.0075 0.0172
0.0006 -0.0001 0.0003 0.0116 -0.0096
-0.0049 -0.0007 -0.0001 -0.0001 -0.6938
-0.0184 0.0099 -0.0033 -0.0004 0.0000
-0.3008 0.0177 0.0143 0.0008 0.0001
0.0032 0.0017 0.0108 -0.0441 -0.0268
5. (1.60474) BD ( 1)Al 2 -Al 4
( 39.74%) 0.6304*Al 2 s( 19.32%)p 4.16( 80.40%)d 0.01( 0.28%)
0.0000 -0.0009 0.4377 0.0330 0.0226
-0.0014 0.0000 -0.0002 0.0495 0.0140
-0.0029 -0.0006 -0.0002 -0.0012 -0.6380
-0.0544 0.0011 -0.0048 -0.0005 -0.0005
-0.6249 -0.0276 -0.0018 -0.0007 -0.0002
0.0093 0.0094 -0.0392 0.0298 -0.0156
( 60.26%) 0.7763*Al 4 s( 16.06%)p 5.21( 83.72%)d 0.01( 0.22%)
0.0000 -0.0001 0.3997 0.0284 0.0008
-0.0010 0.0002 -0.0002 0.0492 0.0068
0.0119 0.0034 -0.0002 -0.0002 0.6527
0.0284 0.0131 0.0018 0.0006 0.0006
0.6384 -0.0048 -0.0066 0.0074 0.0000
-0.0060 -0.0091 -0.0350 0.0126 -0.0266
6. (1.71270) BD ( 1)Al 3 -Al 4
( 50.00%) 0.7071*Al 3 s( 41.20%)p 1.42( 58.60%)d 0.00( 0.21%)
0.0000 -0.0009 0.6415 -0.0201 -0.0011
0.0006 -0.0002 0.0002 0.0336 -0.0031
-0.0131 -0.0043 0.0000 -0.0001 0.2961
0.0150 -0.0009 0.0022 0.0001 -0.0001
0.7047 -0.0064 0.0025 0.0052 -0.0007
0.0009 -0.0026 0.0086 -0.0126 0.0426
( 50.00%) 0.7071*Al 4 s( 41.20%)p 1.42( 58.60%)d 0.00( 0.21%)
0.0000 -0.0009 0.6415 -0.0201 -0.0011
0.0006 -0.0002 0.0002 0.0336 -0.0031
-0.0131 -0.0043 0.0000 -0.0001 0.2961
0.0150 -0.0009 0.0022 0.0001 0.0001
-0.7047 0.0064 -0.0025 -0.0052 0.0007
0.0009 0.0026 -0.0086 -0.0126 0.0426
The occupancy data above suggests the geometry of the Al atoms in the ring is sp2 like however there are clear distortions from this due to the strain produced by the ring. The fact that all the interaction before the non-bonding core s orbital is reached are bonding interactions strongly suggests electrons are being spread across the structure leading to aromaticity.
Analysis of Aluminium D4h pyramidal
Second Order Perturbation Theory Analysis of Fock Matrix in NBO Basis
Threshold for printing: 0.50 kcal/mol
(Intermolecular threshold: 0.05 kcal/mol)
E(2) E(j)-E(i) F(i,j)
Donor NBO (i) Acceptor NBO (j) kcal/mol a.u. a.u.
===================================================================================================
within unit 1
2. BD ( 1)Al 1 -Al 3 / 37. LP ( 1)Al 2 35.49 0.15 0.068
2. BD ( 1)Al 1 -Al 3 / 40. LP ( 1)Al 4 35.49 0.15 0.068
2. BD ( 1)Al 1 -Al 3 /176. BD*( 1)Al 1 -Al 2 33.18 0.28 0.087
2. BD ( 1)Al 1 -Al 3 /178. BD*( 1)Al 1 -Al 4 33.18 0.28 0.087
2. BD ( 1)Al 1 -Al 3 /179. BD*( 1)Al 2 -Al 3 33.18 0.28 0.087
2. BD ( 1)Al 1 -Al 3 /180. BD*( 1)Al 3 -Al 4 33.18 0.28 0.087
5. BD ( 1)Al 3 -Al 4 /177. BD*( 1)Al 1 -Al 3 31.55 0.38 0.101
37. LP ( 1)Al 2 /176. BD*( 1)Al 1 -Al 2 51.13 0.13 0.108
37. LP ( 1)Al 2 /178. BD*( 1)Al 1 -Al 4 19.33 0.13 0.066
37. LP ( 1)Al 2 /179. BD*( 1)Al 2 -Al 3 51.12 0.13 0.108
37. LP ( 1)Al 2 /180. BD*( 1)Al 3 -Al 4 19.33 0.13 0.066
40. LP ( 1)Al 4 /176. BD*( 1)Al 1 -Al 2 19.33 0.13 0.066
40. LP ( 1)Al 4 /178. BD*( 1)Al 1 -Al 4 51.13 0.13 0.108
40. LP ( 1)Al 4 /179. BD*( 1)Al 2 -Al 3 19.33 0.13 0.066
40. LP ( 1)Al 4 /180. BD*( 1)Al 3 -Al 4 51.12 0.13 0.108
It can be seen from the data above that while there is still a large amount of resonance stabilisation in the D4h geometry it is to a lesser extent and there are no interactions above 100kcal/mol like those seen in the Cs geometry. This may explain why the Cs geometry is more thermodynamically stable.
(Occupancy) Bond orbital/ Coefficients/ Hybrids
---------------------------------------------------------------------------------
1. (1.78942) BD ( 1)Al 1 -Al 2
( 43.80%) 0.6618*Al 1 s( 41.30%)p 1.41( 58.33%)d 0.01( 0.37%)
0.0000 -0.0012 0.6422 -0.0076 -0.0216
0.0007 0.0000 0.0003 0.3749 -0.0230
-0.0201 -0.0033 0.0000 -0.0001 0.6604
0.0000 0.0034 0.0058 -0.0008 0.0000
-0.0750 -0.0004 -0.0007 -0.0008 0.0001
0.0134 -0.0019 -0.0112 -0.0541 -0.0219
( 56.20%) 0.7497*Al 2 s( 49.92%)p 1.00( 49.73%)d 0.01( 0.35%)
0.0000 -0.0012 0.7065 0.0021 -0.0100
0.0000 0.0000 0.0004 0.4490 -0.0113
-0.0159 -0.0004 0.0003 0.0001 -0.5383
0.0140 0.0049 -0.0016 0.0008 0.0000
0.0726 -0.0019 -0.0008 0.0002 -0.0001
-0.0010 -0.0006 -0.0084 -0.0579 -0.0056
2. (1.62717) BD ( 1)Al 1 -Al 3
( 50.00%) 0.7071*Al 1 s( 17.58%)p 4.66( 81.97%)d 0.03( 0.44%)
0.0000 -0.0005 0.4173 0.0284 0.0300
-0.0018 0.0001 0.0004 0.5563 0.0246
0.0079 0.0092 0.0003 -0.0005 -0.7066
-0.0312 -0.0101 -0.0117 -0.0004 0.0001
0.0948 0.0042 0.0014 0.0016 0.0001
0.0469 -0.0067 0.0116 0.0111 0.0441
( 50.00%) 0.7071*Al 3 s( 17.58%)p 4.66( 81.97%)d 0.03( 0.44%)
0.0000 -0.0005 0.4173 0.0284 0.0300
-0.0018 0.0001 -0.0004 -0.5563 -0.0246
-0.0079 -0.0092 -0.0003 0.0005 0.7066
0.0313 0.0101 0.0117 0.0004 -0.0001
-0.0948 -0.0042 -0.0014 -0.0016 -0.0001
0.0469 -0.0067 0.0116 0.0111 0.0441
3. (1.78942) BD ( 1)Al 1 -Al 4
( 43.80%) 0.6618*Al 1 s( 41.30%)p 1.41( 58.33%)d 0.01( 0.37%)
0.0000 -0.0012 0.6422 -0.0076 -0.0216
0.0007 0.0000 0.0001 -0.7357 0.0055
0.0015 -0.0049 0.0007 -0.0003 -0.2021
0.0221 0.0202 0.0046 -0.0002 0.0000
0.0135 -0.0026 -0.0024 -0.0006 0.0000
-0.0136 0.0031 0.0016 0.0546 -0.0231
( 56.20%) 0.7497*Al 4 s( 49.92%)p 1.00( 49.73%)d 0.01( 0.35%)
0.0000 -0.0012 0.7065 0.0021 -0.0100
0.0000 0.0000 -0.0002 0.4178 -0.0109
-0.0009 0.0016 -0.0009 -0.0003 -0.5626
0.0142 0.0165 0.0000 -0.0001 0.0000
0.0751 -0.0019 -0.0020 0.0000 0.0000
-0.0280 0.0044 0.0044 0.0509 -0.0069
4. (1.78942) BD ( 1)Al 2 -Al 3
( 56.20%) 0.7497*Al 2 s( 49.92%)p 1.00( 49.73%)d 0.01( 0.35%)
0.0000 -0.0012 0.7065 0.0021 -0.0100
0.0000 0.0000 0.0002 -0.4178 0.0109
0.0009 -0.0016 0.0009 0.0003 0.5626
-0.0142 -0.0165 0.0000 0.0001 0.0000
-0.0751 0.0019 0.0020 0.0000 0.0000
-0.0280 0.0044 0.0044 0.0509 -0.0069
( 43.80%) 0.6618*Al 3 s( 41.30%)p 1.41( 58.33%)d 0.01( 0.37%)
0.0000 -0.0012 0.6422 -0.0076 -0.0216
0.0007 0.0000 -0.0001 0.7357 -0.0055
-0.0015 0.0049 -0.0007 0.0003 0.2021
-0.0221 -0.0202 -0.0046 0.0002 0.0000
-0.0135 0.0026 0.0024 0.0006 0.0000
-0.0136 0.0031 0.0016 0.0546 -0.0231
5. (1.78942) BD ( 1)Al 3 -Al 4
( 43.80%) 0.6618*Al 3 s( 41.30%)p 1.41( 58.33%)d 0.01( 0.37%)
0.0000 -0.0012 0.6422 -0.0076 -0.0216
0.0007 0.0000 -0.0003 -0.3749 0.0230
0.0201 0.0033 0.0000 0.0001 -0.6604
0.0000 -0.0034 -0.0058 0.0008 0.0000
0.0750 0.0004 0.0007 0.0008 -0.0001
0.0134 -0.0019 -0.0112 -0.0541 -0.0219
( 56.20%) 0.7497*Al 4 s( 49.92%)p 1.00( 49.73%)d 0.01( 0.35%)
0.0000 -0.0012 0.7065 0.0021 -0.0100
0.0000 0.0000 -0.0004 -0.4490 0.0113
0.0159 0.0004 -0.0003 -0.0001 0.5383
-0.0140 -0.0049 0.0016 -0.0008 0.0000
-0.0726 0.0019 0.0008 -0.0002 0.0001
-0.0010 -0.0006 -0.0084 -0.0579 -0.0056
6. (2.00000) CR ( 1)Al 1 s(100.00%)
1.0000 0.0000 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.0000 0.0000
The bonding in the D4h structure is very similar to that in the Cs geometry, again orbitals 1 to 5 are all bonding interactions and this suggests aromaticity.
Analysis of Gallium Cs
Second Order Perturbation Theory Analysis of Fock Matrix in NBO Basis
Threshold for printing: 0.50 kcal/mol
(Intermolecular threshold: 0.05 kcal/mol)
E(2) E(j)-E(i) F(i,j)
Donor NBO (i) Acceptor NBO (j) kcal/mol a.u. a.u.
===================================================================================================
within unit 1
72. LP*( 1)Na 1 / 73. LP*( 2)Na 1 18.83 0.09 0.085
from unit 1 to unit 2
72. LP*( 1)Na 1 / 76. LP*( 1)Na 2 24.01 0.09 0.081
from unit 1 to unit 3
from unit 2 to unit 1
within unit 2
76. LP*( 1)Na 2 / 78. LP*( 3)Na 2 141.66 0.02 0.160
from unit 2 to unit 3
from unit 3 to unit 1
80. LP ( 1)Ga 3 / 72. LP*( 1)Na 1 37.31 0.06 0.064
82. LP*( 2)Ga 4 / 72. LP*( 1)Na 1 42.37 0.03 0.055
84. LP*( 2)Ga 5 / 72. LP*( 1)Na 1 37.00 0.06 0.063
85. LP*( 1)Ga 6 / 72. LP*( 1)Na 1 44.19 0.03 0.054
from unit 3 to unit 2
2. BD ( 1)Ga 3 -Ga 5 / 76. LP*( 1)Na 2 34.48 0.65 0.136
252. BD*( 1)Ga 5 -Ga 6 / 76. LP*( 1)Na 2 28.81 0.02 0.040
within unit 3
1. BD ( 1)Ga 3 -Ga 4 /250. BD*( 1)Ga 3 -Ga 6 28.91 0.42 0.104
1. BD ( 1)Ga 3 -Ga 4 /251. BD*( 1)Ga 4 -Ga 6 21.12 0.63 0.106
2. BD ( 1)Ga 3 -Ga 5 / 81. LP*( 1)Ga 4 17.67 0.52 0.090
2. BD ( 1)Ga 3 -Ga 5 /250. BD*( 1)Ga 3 -Ga 6 42.28 0.40 0.122
3. BD ( 1)Ga 3 -Ga 6 / 81. LP*( 1)Ga 4 19.59 0.19 0.055
3. BD ( 1)Ga 3 -Ga 6 / 83. LP ( 1)Ga 5 22.65 0.15 0.055
3. BD ( 1)Ga 3 -Ga 6 /248. BD*( 1)Ga 3 -Ga 4 37.81 0.28 0.093
3. BD ( 1)Ga 3 -Ga 6 /249. BD*( 1)Ga 3 -Ga 5 78.88 0.22 0.118
3. BD ( 1)Ga 3 -Ga 6 /251. BD*( 1)Ga 4 -Ga 6 39.92 0.29 0.099
3. BD ( 1)Ga 3 -Ga 6 /252. BD*( 1)Ga 5 -Ga 6 36.87 0.31 0.100
4. BD ( 1)Ga 4 -Ga 6 /248. BD*( 1)Ga 3 -Ga 4 19.85 0.57 0.097
4. BD ( 1)Ga 4 -Ga 6 /250. BD*( 1)Ga 3 -Ga 6 55.67 0.37 0.133
60. CR ( 3)Ga 6 /250. BD*( 1)Ga 3 -Ga 6 29.11 8.16 0.489
80. LP ( 1)Ga 3 / 82. LP*( 2)Ga 4 148.46 0.03 0.085
81. LP*( 1)Ga 4 /248. BD*( 1)Ga 3 -Ga 4 91.71 0.09 0.126
81. LP*( 1)Ga 4 /249. BD*( 1)Ga 3 -Ga 5 117.47 0.03 0.077
83. LP ( 1)Ga 5 / 81. LP*( 1)Ga 4 99.59 0.04 0.077
83. LP ( 1)Ga 5 /249. BD*( 1)Ga 3 -Ga 5 196.70 0.07 0.139
84. LP*( 2)Ga 5 / 85. LP*( 1)Ga 6 138.19 0.03 0.084
248. BD*( 1)Ga 3 -Ga 4 /251. BD*( 1)Ga 4 -Ga 6 373.00 0.01 0.108
249. BD*( 1)Ga 3 -Ga 5 /248. BD*( 1)Ga 3 -Ga 4 89.97 0.06 0.101
249. BD*( 1)Ga 3 -Ga 5 /252. BD*( 1)Ga 5 -Ga 6 45.63 0.09 0.097
251. BD*( 1)Ga 4 -Ga 6 /252. BD*( 1)Ga 5 -Ga 6 135.75 0.02 0.093
It can be seen from stabilisation energies above that with Gallium the resonances are very high, even higher than those found in aluminium. However it should be taken into consideration that Gallium is larger than aluminium and therefore had larger orbitals, so these larger resonances are to be expected.
(Occupancy) Bond orbital/ Coefficients/ Hybrids
---------------------------------------------------------------------------------
1. (1.81751) BD ( 1)Ga 3 -Ga 4
( 45.04%) 0.6711*Ga 3 s( 41.62%)p 1.39( 58.03%)d 0.01( 0.34%)
0.0000 0.0000 -0.0009 0.6451 -0.0058
-0.0063 -0.0011 0.0001 0.0000 0.0000
0.0002 0.0030 0.0116 0.0057 -0.0008
-0.0003 0.0000 0.0000 -0.0001 -0.7065
0.0209 0.0018 -0.0015 -0.0009 0.0001
0.0000 0.0000 0.2833 0.0171 0.0086
-0.0007 0.0000 0.0000 -0.0002 0.0065
-0.0009 0.0002 -0.0004 -0.0005 -0.0004
0.0085 -0.0050 -0.0002 -0.0497 -0.0050
0.0003 -0.0279 -0.0034
( 54.96%) 0.7414*Ga 4 s( 53.22%)p 0.87( 46.47%)d 0.01( 0.32%)
0.0000 0.0000 -0.0010 0.7293 -0.0162
-0.0036 -0.0008 0.0000 0.0000 0.0000
0.0002 0.0124 0.0067 0.0057 0.0011
0.0008 0.0000 0.0000 0.0000 0.4258
0.0051 -0.0010 0.0028 0.0025 0.0000
0.0000 0.0005 0.5321 0.0065 0.0017
0.0005 -0.0007 0.0000 0.0001 -0.0050
0.0008 0.0002 -0.0056 0.0002 0.0002
0.0127 0.0017 -0.0003 -0.0308 -0.0017
-0.0001 -0.0445 -0.0029
2. (1.75387) BD ( 1)Ga 3 -Ga 5
( 39.63%) 0.6295*Ga 3 s( 39.59%)p 1.52( 60.04%)d 0.01( 0.37%)
0.0000 0.0000 -0.0011 0.6291 -0.0126
-0.0007 -0.0005 0.0004 0.0000 0.0000
0.0001 0.0421 0.0085 0.0055 -0.0018
0.0000 0.0000 0.0000 -0.0001 0.2871
-0.0094 -0.0024 0.0010 0.0009 0.0000
0.0000 0.0002 -0.7183 0.0058 0.0027
0.0002 -0.0020 0.0000 0.0002 0.0061
-0.0010 -0.0002 0.0061 -0.0012 -0.0004
-0.0031 -0.0013 0.0004 -0.0331 0.0021
0.0008 0.0503 0.0027
( 60.37%) 0.7770*Ga 5 s( 50.18%)p 0.99( 49.59%)d 0.00( 0.23%)
0.0000 0.0000 -0.0012 0.7083 -0.0046
-0.0003 0.0003 0.0002 0.0000 0.0000
0.0001 0.0612 0.0016 0.0050 -0.0023
0.0003 0.0000 0.0000 -0.0001 0.5482
-0.0233 -0.0034 0.0029 0.0000 0.0000
0.0000 -0.0003 0.4370 -0.0070 -0.0025
-0.0025 0.0015 0.0000 0.0002 0.0017
-0.0004 0.0001 0.0011 0.0004 0.0004
0.0281 -0.0007 0.0005 -0.0194 0.0028
0.0009 0.0332 0.0019
3. (1.57277) BD ( 1)Ga 3 -Ga 6
( 58.50%) 0.7648*Ga 3 s( 18.24%)p 4.45( 81.10%)d 0.04( 0.66%)
0.0000 0.0000 -0.0007 0.4267 0.0175
0.0001 0.0020 -0.0005 0.0000 0.0000
-0.0002 0.0589 -0.0145 0.0005 -0.0019
0.0006 0.0000 0.0000 -0.0001 0.6402
-0.0351 -0.0028 0.0050 -0.0022 0.0000
0.0000 0.0005 0.6294 0.0068 0.0013
0.0060 0.0010 0.0000 0.0000 -0.0094
0.0011 0.0002 -0.0177 0.0016 -0.0004
-0.0647 0.0043 0.0001 0.0219 0.0015
0.0005 -0.0384 -0.0021
( 41.50%) 0.6442*Ga 6 s( 21.25%)p 3.67( 78.02%)d 0.03( 0.73%)
0.0000 0.0000 -0.0011 0.4600 0.0293
-0.0061 -0.0010 -0.0004 0.0000 0.0000
-0.0002 0.0609 -0.0193 0.0046 -0.0008
0.0015 0.0000 0.0000 -0.0012 -0.6286
0.0449 0.0025 -0.0084 0.0009 0.0000
0.0000 -0.0004 -0.6151 0.0172 0.0093
-0.0013 0.0021 0.0000 -0.0003 0.0155
-0.0018 -0.0001 0.0174 -0.0020 0.0000
-0.0571 0.0043 0.0002 0.0530 0.0017
0.0001 -0.0260 -0.0023
4. (1.75040) BD ( 1)Ga 4 -Ga 6
( 55.72%) 0.7464*Ga 4 s( 46.65%)p 1.14( 52.98%)d 0.01( 0.38%)
0.0000 0.0000 -0.0013 0.6829 0.0116
0.0018 0.0014 0.0001 0.0000 0.0000
0.0003 -0.0027 0.0010 0.0021 0.0022
0.0003 0.0000 0.0000 -0.0005 -0.4926
0.0023 -0.0018 -0.0044 0.0000 0.0000
0.0000 0.0001 -0.5358 -0.0034 0.0025
0.0019 -0.0010 0.0000 -0.0002 0.0038
0.0001 -0.0002 0.0080 -0.0010 0.0003
0.0125 0.0016 -0.0001 0.0196 0.0020
0.0003 0.0559 0.0041
( 44.28%) 0.6655*Ga 6 s( 37.34%)p 1.67( 62.22%)d 0.01( 0.45%)
0.0000 0.0000 -0.0011 0.6110 0.0013
0.0037 0.0017 0.0002 0.0000 0.0000
0.0003 -0.0225 0.0080 0.0010 0.0027
-0.0001 0.0000 0.0000 -0.0003 -0.2806
-0.0123 -0.0028 -0.0017 -0.0001 0.0000
0.0000 0.0000 0.7366 -0.0026 -0.0059
-0.0016 0.0004 -0.0001 -0.0002 -0.0016
0.0008 0.0002 -0.0139 0.0017 -0.0003
0.0052 -0.0032 -0.0002 0.0008 0.0014
0.0003 0.0648 0.0049
5. (1.76421) BD ( 1)Ga 5 -Ga 6
( 58.23%) 0.7631*Ga 5 s( 49.18%)p 1.03( 50.48%)d 0.01( 0.33%)
0.0000 0.0000 -0.0009 0.7013 -0.0007
-0.0060 -0.0005 0.0000 0.0000 0.0000
0.0002 0.0156 0.0073 0.0053 -0.0011
-0.0001 0.0000 0.0000 -0.0001 -0.5372
0.0119 0.0012 -0.0003 -0.0014 0.0001
0.0000 -0.0001 -0.4641 -0.0180 -0.0086
-0.0008 -0.0004 0.0000 -0.0002 0.0036
-0.0006 -0.0002 0.0050 0.0001 0.0004
0.0077 0.0038 -0.0002 -0.0411 -0.0046
0.0003 -0.0389 -0.0039
( 41.77%) 0.6463*Ga 6 s( 41.53%)p 1.40( 58.01%)d 0.01( 0.46%)
0.0000 0.0000 -0.0009 0.6437 -0.0302
-0.0011 -0.0003 0.0002 0.0000 0.0000
0.0003 -0.0145 0.0163 0.0043 0.0017
0.0002 0.0000 0.0000 0.0003 0.7153
-0.0147 -0.0024 0.0066 0.0023 0.0000
0.0000 -0.0005 -0.2596 -0.0148 -0.0064
-0.0002 -0.0001 0.0000 0.0002 -0.0123
0.0017 -0.0002 -0.0024 0.0008 -0.0002
0.0112 -0.0039 -0.0003 -0.0565 -0.0025
-0.0001 -0.0325 -0.0018
6. (2.00000) CR ( 1)Na 1 s(100.00%)
1.0000 0.0000 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.0000 0.0000
0.0000 0.0000 0.0000 0.0000 0.0000
The bonding picture for the Gallium geometries is again the same as in the Aluminium molecules.
Vibrational Spectroscopy Analysis
Frequency analysis was run simply to check that the optimisations run previously had reached global minima. Presence of the low frequency absoprtions close to zero suggests this is the case.
An example for Aluminium in the Cs geometry is shown below:
Full mass-weighted force constant matrix: Low frequencies --- -1.8179 -1.8125 -0.0020 0.0006 0.0018 16.9290 Low frequencies --- 68.8340 68.8341 84.5201
Conclusion of Mini Project
In conclusion the experiment has been successful in illustrating that it is possible for all metal clusters to have both π and σ aromaticity. In addition the geometry analyses has shown that the Cs geometry is the most stable of the ones investigated. NBO analysis has suggested the reason for this due to greater σ aromaticity between the metal atoms.
Conclusion
Computational techniques have been shown to be very useful in the calculation of almost all aspects of inorganic molecules despite their often unusual bonding. Geometry optimisations using the appropriate basis set have produced geometries with relative energies that agree well with literature. From these geometries it has been seen than IR spectra can be predicted to a good accuracy and molecular orbitals and NBO analysis provide an insight into the nature of where electrons are located in complex molecules which is simply not accessible by experiments in the wet lab.
References
- ↑ Kentarou Kawaguchi, J. E. Butler, Chikashi Yamada, S. H. Bauer, Tatsuya Minowa, Hideto Kanamori, and Eizi Hirota , J. Chem. Phys. 87, 2438 (1987). http://dx.doi.org/10.1063/1.453135
- ↑ http://hdl.handle.net/10042/to-12085
- ↑ http://en.wikipedia.org/wiki/Electronegativity
- ↑ http://hdl.handle.net/10042/to-12086
- ↑ Johan Blixt, Julius Glaser, Janos Mink, Ingmar Persson, Per Persson, Magnus Sandstroem, J. Am. Chem. Soc., 1995, 117 (18), pp 5089–5104. http://pubs.acs.org/doi/abs/10.1021/ja00123a011
- ↑ http://hdl.handle.net/10042/to-12087
- ↑ http://hdl.handle.net/10042/to-12088
- ↑ http://hdl.handle.net/10042/to-12089
- ↑ http://hdl.handle.net/10042/to-12090
- ↑ http://hdl.handle.net/10042/to-12091
- ↑ http://hdl.handle.net/10042/to-12092
- ↑ F. Albert. Cotton, Donald J. Darensbourg, Simonetta. Klein, Brian W. S. Kolthammer. Inorg. Chem., 1982, 21 (4), pp 1651–1655 http://pubs.acs.org/doi/pdf/10.1021/ic00134a075
- ↑ D. W. Bennett, J. Chem. Cryst., 34(6), 2004, pp 353 - 359. http://www.springerlink.com/content/u5383x017700g761/fulltext.pdf
- ↑ http://mccammon.ucsd.edu/~dzhang/energy-unit-conv-table.html
- ↑ 3rd Year Computational Chemistry Online Manual, Module 2, 2011
- ↑ http://hdl.handle.net/10042/to-12093
- ↑ http://hdl.handle.net/10042/to-12094
- ↑ Florence M. Nareetsile and N.J. Coville, Bull. Chem. Soc. Ethiop. 2011, 25(2). http://www.ajol.info/index.php/bcse/article/viewFile/65884/53602
- ↑ Anthony W. Brown and Anthony L. Diaz, J. Chem. Educ., 2010, 87 (9), pp 975–977. http://pubs.acs.org/doi/pdf/10.1021/ed1002477
- ↑ Fowler, R.W.A. Havenith, E. Steine Chemical Physics Letters Volume 359, Issues 5–6, 27 June 2002, Pages 530–536
- ↑ Alexander I. Boldyrev* and Aleksey E. Kuznetsov, Inorg. Chem., 2002, 41 (3), pp 532–53 DOI: 10.1021/ic010840u
- ↑ http://hdl.handle.net/10042/to-12103
- ↑ http://hdl.handle.net/10042/to-12095
- ↑ http://hdl.handle.net/10042/to-12112
- ↑ http://hdl.handle.net/10042/to-12111
- ↑ http://hdl.handle.net/10042/to-12127
- ↑ http://hdl.handle.net/10042/to-12097
- ↑ http://hdl.handle.net/10042/to-12096
- ↑ http://hdl.handle.net/10042/to-12116
- ↑ http://hdl.handle.net/10042/to-12117
- ↑ http://hdl.handle.net/10042/to-12102
- ↑ http://hdl.handle.net/10042/to-12099
- ↑ http://hdl.handle.net/10042/to-12101
- ↑ http://hdl.handle.net/10042/to-12104
- ↑ http://hdl.handle.net/10042/to-12107
- ↑ http://hdl.handle.net/10042/to-12115
- ↑ http://hdl.handle.net/10042/to-12114
- ↑ http://hdl.handle.net/10042/to-12128
- ↑ http://hdl.handle.net/10042/to-12105
- ↑ http://hdl.handle.net/10042/to-12106
- ↑ http://hdl.handle.net/10042/to-12118
- ↑ http://hdl.handle.net/10042/to-12113
- ↑ http://hdl.handle.net/10042/to-12109
- ↑ http://hdl.handle.net/10042/to-12108
- ↑ http://hdl.handle.net/10042/to-12110
- ↑ http://hdl.handle.net/10042/to-12120
- ↑ http://hdl.handle.net/10042/to-12098
- ↑ http://hdl.handle.net/10042/to-12119
- ↑ http://hdl.handle.net/10042/to-12122
- ↑ http://hdl.handle.net/10042/to-12121
- ↑ http://hdl.handle.net/10042/to-12124
- ↑ http://hdl.handle.net/10042/to-12123
- ↑ http://hdl.handle.net/10042/to-12110
- ↑ http://hdl.handle.net/10042/to-12126
- ↑ http://hdl.handle.net/10042/to-12125
- ↑ Constantinos A. Tsipis, Coordination Chemistry Reviews, Volume 249, Issue 24, 15 December 2005, Pages 2740–2762